

High-resolution profiling of pathways of escape for SARS-CoV-2 spike-binding antibodies
- PMID: 34010620
- PMCID: PMC8096189
- DOI: 10.1016/j.cell.2021.04.045
High-resolution profiling of pathways of escape for SARS-CoV-2 spike-binding antibodies
Abstract
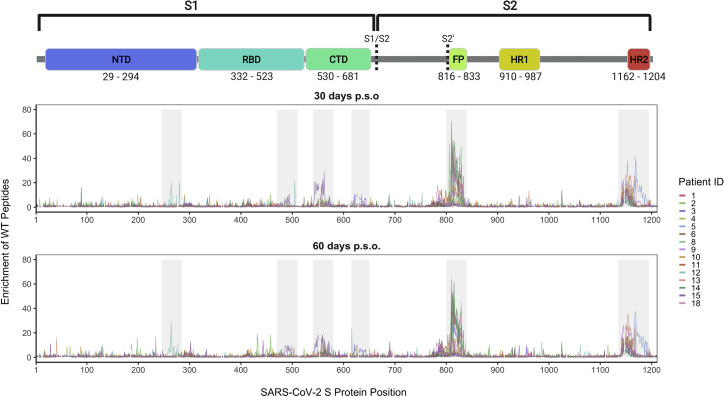
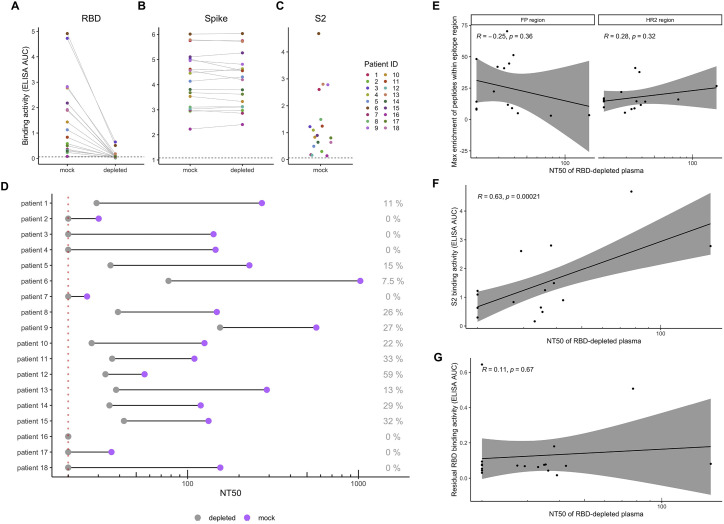
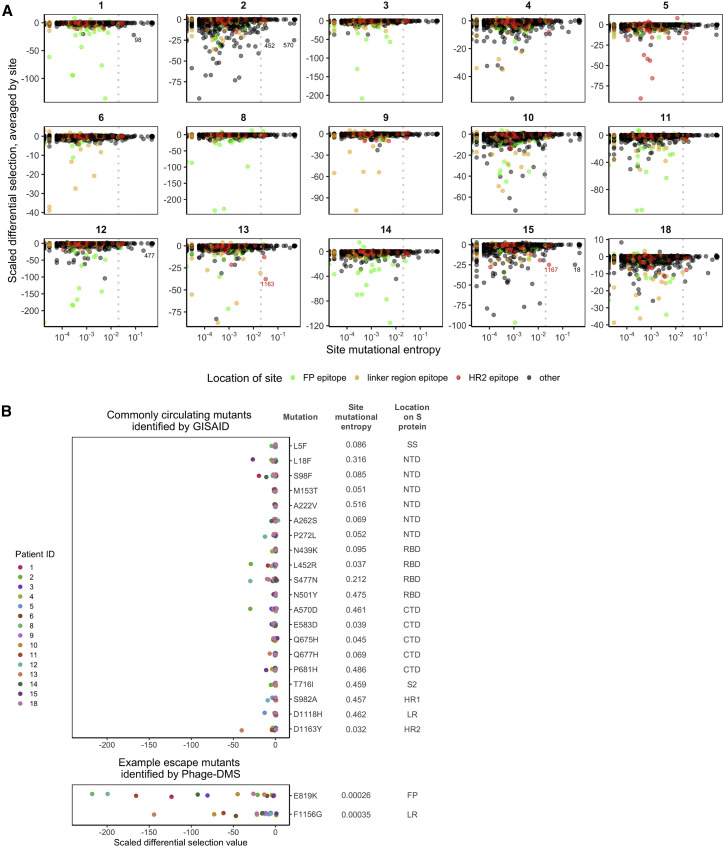
Defining long-term protective immunity to SARS-CoV-2 is one of the most pressing questions of our time and will require a detailed understanding of potential ways this virus can evolve to escape immune protection. Immune protection will most likely be mediated by antibodies that bind to the viral entry protein, spike (S). Here, we used Phage-DMS, an approach that comprehensively interrogates the effect of all possible mutations on binding to a protein of interest, to define the profile of antibody escape to the SARS-CoV-2 S protein using coronavirus disease 2019 (COVID-19) convalescent plasma. Antibody binding was common in two regions, the fusion peptide and the linker region upstream of the heptad repeat region 2. However, escape mutations were variable within these immunodominant regions. There was also individual variation in less commonly targeted epitopes. This study provides a granular view of potential antibody escape pathways and suggests there will be individual variation in antibody-mediated virus evolution.
Keywords: COVID-19; DMS; SARS-CoV-2; Spike; antibodies; deep mutational scanning; epitope mapping; escape mutations; phage display.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.E.G. and J.O. are inventors on a patent application on Phage-DMS. H.Y.C. is a consultant for Merck, Pfizer, Ellume, and the Bill and Melinda Gates Foundation and has received support from Cepheid and Sanofi-Pasteur.
Figures










Update of
-
High resolution profiling of pathways of escape for SARS-CoV-2 spike-binding antibodies.bioRxiv [Preprint]. 2020 Nov 16:2020.11.16.385278. doi: 10.1101/2020.11.16.385278. bioRxiv. 2020. Update in: Cell. 2021 May 27;184(11):2927-2938.e11. doi: 10.1016/j.cell.2021.04.045. PMID: 33236010 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
