

Human telomerase is directly regulated by non-telomeric TRF2-G-quadruplex interaction
- PMID: 34010660
- PMCID: PMC7611063
- DOI: 10.1016/j.celrep.2021.109154
Human telomerase is directly regulated by non-telomeric TRF2-G-quadruplex interaction
Abstract
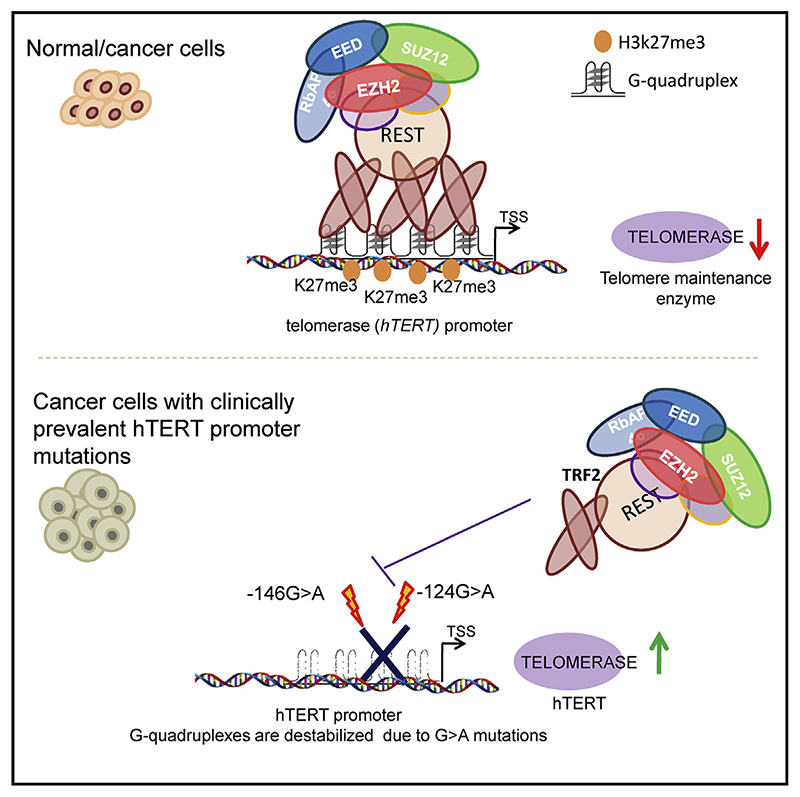
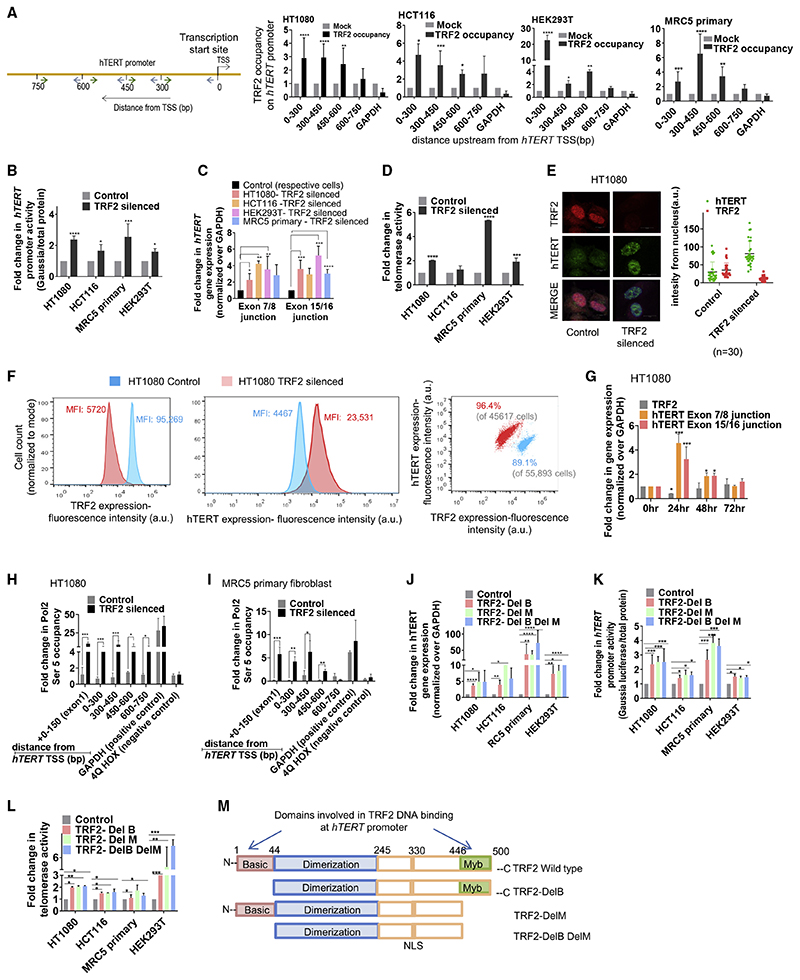
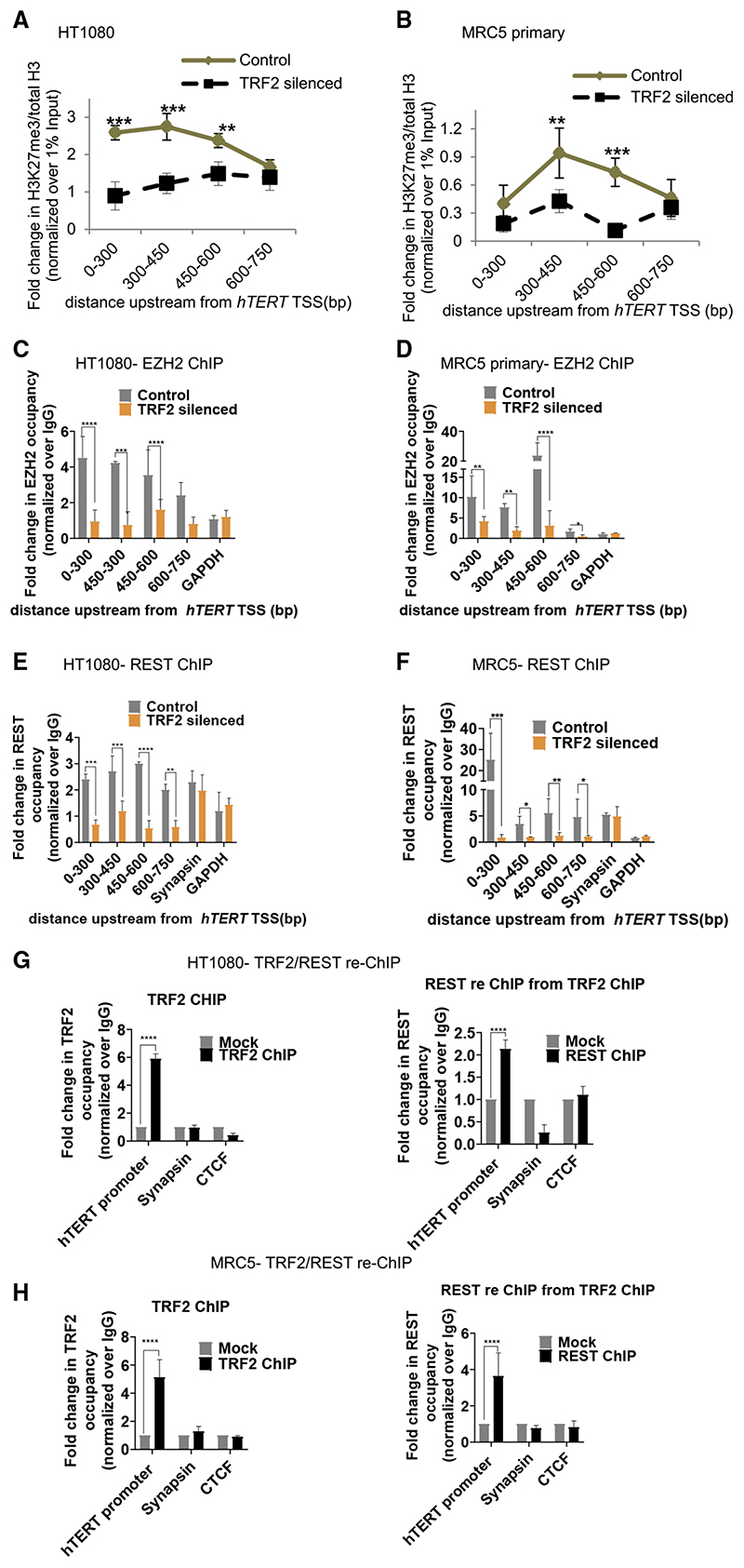
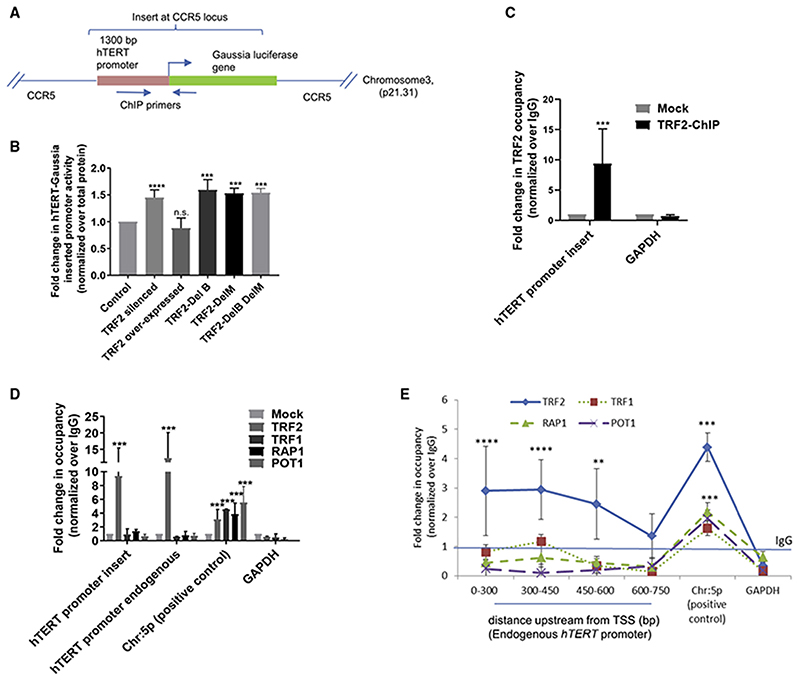
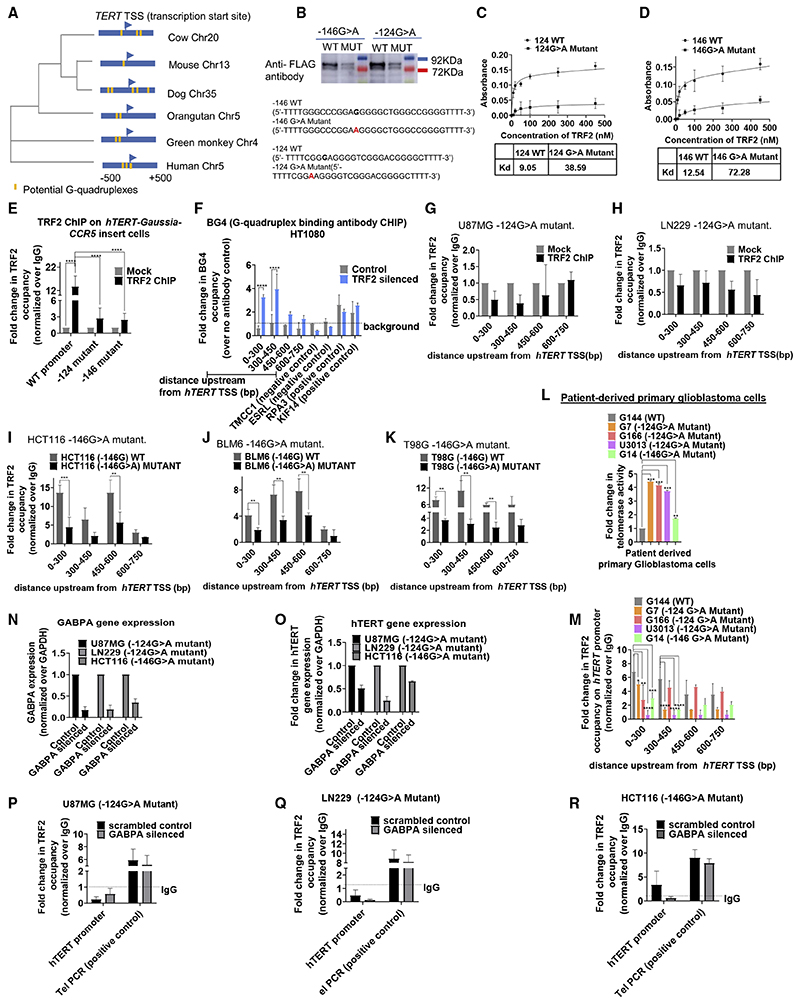
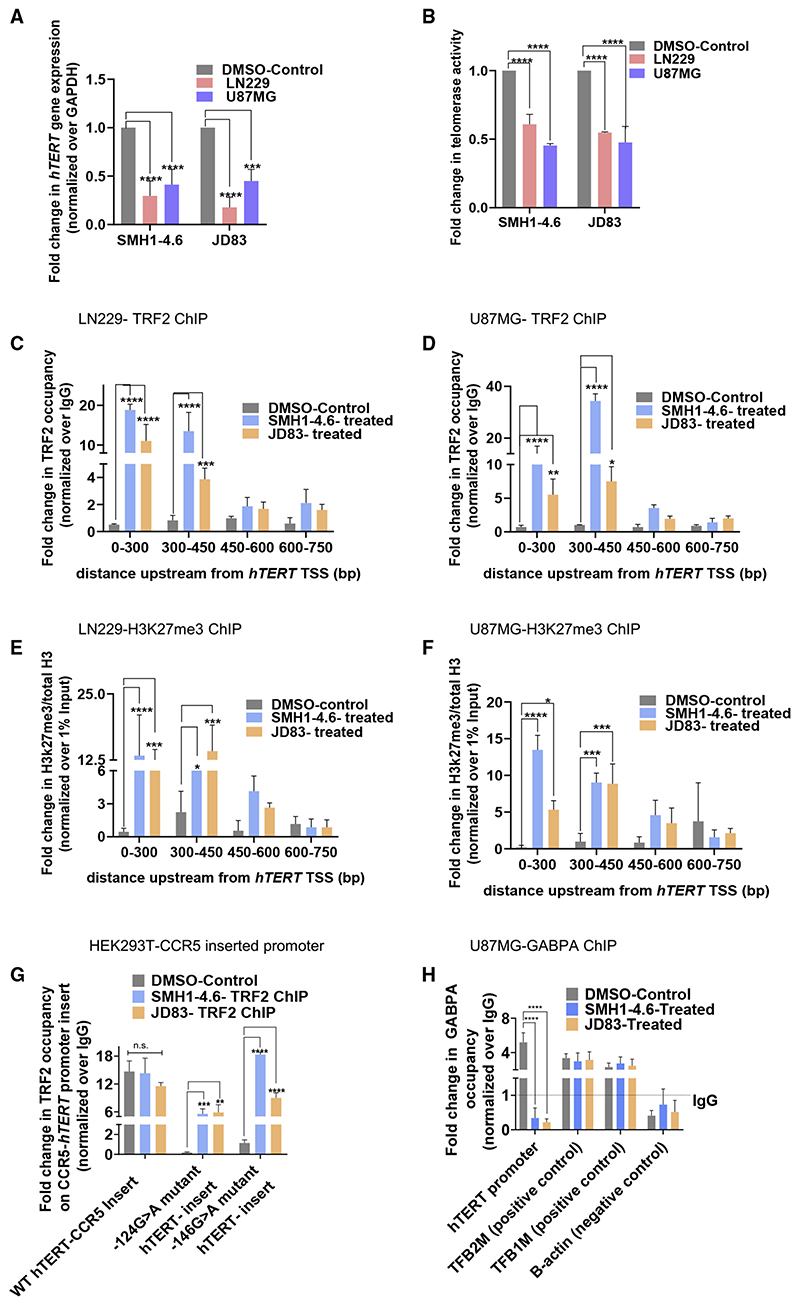
Human telomerase reverse transcriptase (hTERT) remains suppressed in most normal somatic cells. Resulting erosion of telomeres leads eventually to replicative senescence. Reactivation of hTERT maintains telomeres and triggers progression of >90% of cancers. However, any direct causal link between telomeres and telomerase regulation remains unclear. Here, we show that the telomere-repeat-binding-factor 2 (TRF2) binds hTERT promoter G-quadruplexes and recruits the polycomb-repressor EZH2/PRC2 complex. This is causal for H3K27 trimethylation at the hTERT promoter and represses hTERT in cancer as well as normal cells. Two highly recurrent hTERT promoter mutations found in many cancers, including ∼83% glioblastoma multiforme, that are known to destabilize hTERT promoter G-quadruplexes, showed loss of TRF2 binding in patient-derived primary glioblastoma multiforme cells. Ligand-induced G-quadruplex stabilization restored TRF2 binding, H3K27-trimethylation, and hTERT re-suppression. These results uncover a mechanism of hTERT regulation through a telomeric factor, implicating telomere-telomerase molecular links important in neoplastic transformation, aging, and regenerative therapy.
Keywords: G-quadruplexe; PRC2; REST; cancer; epigentics; glioblastoma; hTERT promoter mutations; shelterin proteins; telomerase; telomere repeat binding factor.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.C. is a scientific advisor for Rejuvas Biotech India Pvt. Ltd.
Figures
References
-
- Aguado T, Gutiérrez FJ, Aix E, Schneider RP, Giovinazzo G, Blasco MA, Flores I. Telomere Length Defines the Cardiomyocyte Differentiation Potency of Mouse Induced Pluripotent Stem Cells. Stem Cells. 2017;35:362–373. - PubMed
-
- Ak?nc?lar SC, Khattar E, Boon PLS, Unal B, Fullwood MJ, Ter-gaonkar V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016;6:1276–1291. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
