

An inter-laboratory study to investigate the impact of the bioinformatics component on microbiome analysis using mock communities
- PMID: 34012005
- PMCID: PMC8134577
- DOI: 10.1038/s41598-021-89881-2
An inter-laboratory study to investigate the impact of the bioinformatics component on microbiome analysis using mock communities
Abstract
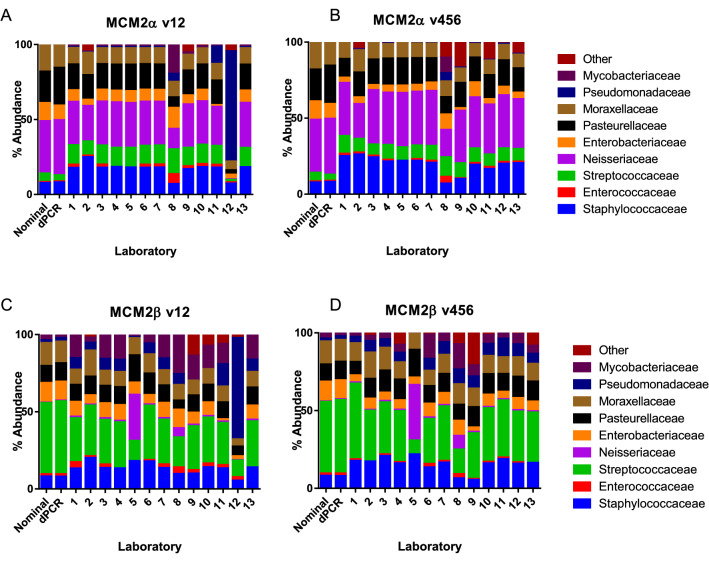
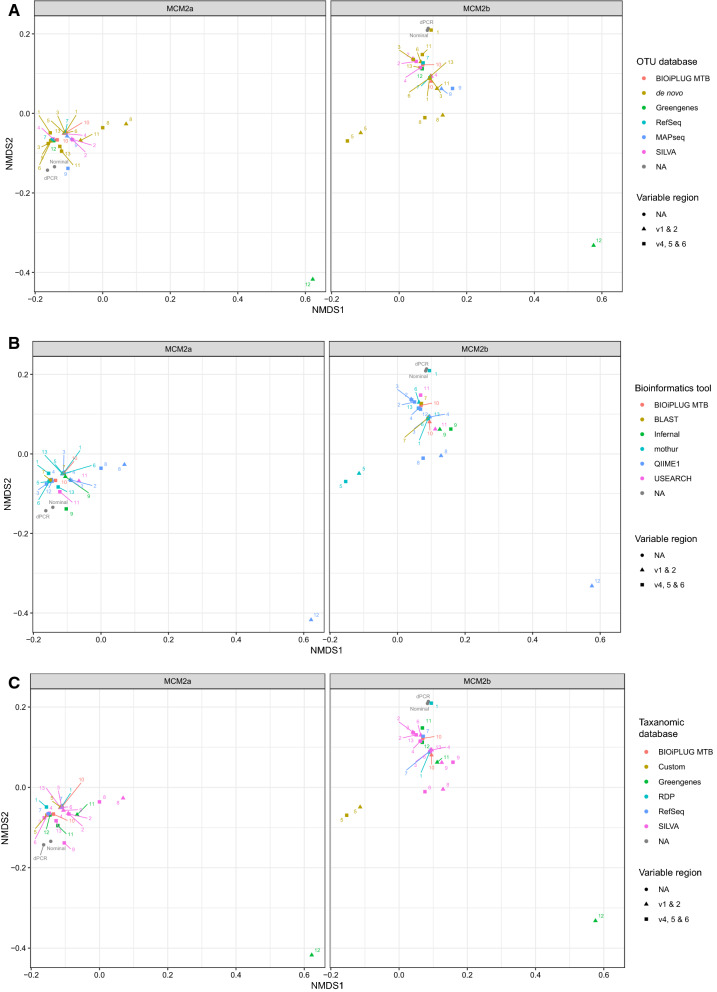
Despite the advent of whole genome metagenomics, targeted approaches (such as 16S rRNA gene amplicon sequencing) continue to be valuable for determining the microbial composition of samples. Amplicon microbiome sequencing can be performed on clinical samples from a normally sterile site to determine the aetiology of an infection (usually single pathogen identification) or samples from more complex niches such as human mucosa or environmental samples where multiple microorganisms need to be identified. The methodologies are frequently applied to determine both presence of micro-organisms and their quantity or relative abundance. There are a number of technical steps required to perform microbial community profiling, many of which may have appreciable precision and bias that impacts final results. In order for these methods to be applied with the greatest accuracy, comparative studies across different laboratories are warranted. In this study we explored the impact of the bioinformatic approaches taken in different laboratories on microbiome assessment using 16S rRNA gene amplicon sequencing results. Data were generated from two mock microbial community samples which were amplified using primer sets spanning five different variable regions of 16S rRNA genes. The PCR-sequencing analysis included three technical repeats of the process to determine the repeatability of their methods. Thirteen laboratories participated in the study, and each analysed the same FASTQ files using their choice of pipeline. This study captured the methods used and the resulting sequence annotation and relative abundance output from bioinformatic analyses. Results were compared to digital PCR assessment of the absolute abundance of each target representing each organism in the mock microbial community samples and also to analyses of shotgun metagenome sequence data. This ring trial demonstrates that the choice of bioinformatic analysis pipeline alone can result in different estimations of the composition of the microbiome when using 16S rRNA gene amplicon sequencing data. The study observed differences in terms of both presence and abundance of organisms and provides a resource for ensuring reproducible pipeline development and application. The observed differences were especially prevalent when using custom databases and applying high stringency operational taxonomic unit (OTU) cut-off limits. In order to apply sequencing approaches with greater accuracy, the impact of different analytical steps needs to be clearly delineated and solutions devised to harmonise microbiome analysis results.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Exploring animal food microbiomes and resistomes via 16S rRNA gene amplicon sequencing and shotgun metagenomics.Appl Environ Microbiol. 2025 Feb 19;91(2):e0223024. doi: 10.1128/aem.02230-24. Epub 2025 Jan 22. Appl Environ Microbiol. 2025. PMID: 39840975 Free PMC article.
-
Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome.OMICS. 2018 Apr;22(4):248-254. doi: 10.1089/omi.2018.0013. OMICS. 2018. PMID: 29652573
-
Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing.mSphere. 2021 Feb 24;6(1):e01202-20. doi: 10.1128/mSphere.01202-20. mSphere. 2021. PMID: 33627512 Free PMC article.
-
Metatranscriptomics: A Tool for Clinical Metagenomics.OMICS. 2024 Aug;28(8):394-407. doi: 10.1089/omi.2024.0130. Epub 2024 Jul 19. OMICS. 2024. PMID: 39029911 Review.
-
Critical review of 16S rRNA gene sequencing workflow in microbiome studies: From primer selection to advanced data analysis.Mol Oral Microbiol. 2023 Oct;38(5):347-399. doi: 10.1111/omi.12434. Epub 2023 Oct 7. Mol Oral Microbiol. 2023. PMID: 37804481 Review.
Cited by
-
Metrological framework to support accurate, reliable, and reproducible nucleic acid measurements.Anal Bioanal Chem. 2022 Jan;414(2):791-806. doi: 10.1007/s00216-021-03712-x. Epub 2021 Nov 4. Anal Bioanal Chem. 2022. PMID: 34738220 Free PMC article. Review.
-
Standardization of 16S rRNA gene sequencing using nanopore long read sequencing technology for clinical diagnosis of culture negative infections.Front Cell Infect Microbiol. 2025 Mar 6;15:1517208. doi: 10.3389/fcimb.2025.1517208. eCollection 2025. Front Cell Infect Microbiol. 2025. PMID: 40115075 Free PMC article.
-
Multi-Omics Strategies for Investigating the Microbiome in Toxicology Research.Toxicol Sci. 2022 May 26;187(2):189-213. doi: 10.1093/toxsci/kfac029. Toxicol Sci. 2022. PMID: 35285497 Free PMC article. Review.
-
Reducing bias in microbiome research: Comparing methods from sample collection to sequencing.Front Microbiol. 2023 Mar 30;14:1094800. doi: 10.3389/fmicb.2023.1094800. eCollection 2023. Front Microbiol. 2023. PMID: 37065158 Free PMC article.
-
Using 16s rRNA sequencing to characterize the microbiome of tropical cutaneous ulcer disease: insights into the microbial landscape and implications for diagnosis and treatment.Microb Genom. 2024 May;10(5):001234. doi: 10.1099/mgen.0.001234. Microb Genom. 2024. PMID: 38739120 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- BBS/E/F/000PR10349/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/N010469/1/MRC UK
- MR/N010469/1/MRC_/Medical Research Council/United Kingdom
- BBS/E/F/000PR10348/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/K000551/1/MRC UK
- MR/N013956/1/MRC_/Medical Research Council/United Kingdom
- MR/R020973/1/MRC UK
- BB/R012504/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/R020973/1/MRC_/Medical Research Council/United Kingdom
- MR/M01360X/1/MRC UK
- BB/R013063/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/N013956/1/UK Antimicrobial Resistance Cross Council Initiative
- MR/L015080/1/MRC_/Medical Research Council/United Kingdom
- MR/M01360X/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
