

MIR-NATs repress MAPT translation and aid proteostasis in neurodegeneration
- PMID: 34012113
- PMCID: PMC7610982
- DOI: 10.1038/s41586-021-03556-6
MIR-NATs repress MAPT translation and aid proteostasis in neurodegeneration
Abstract
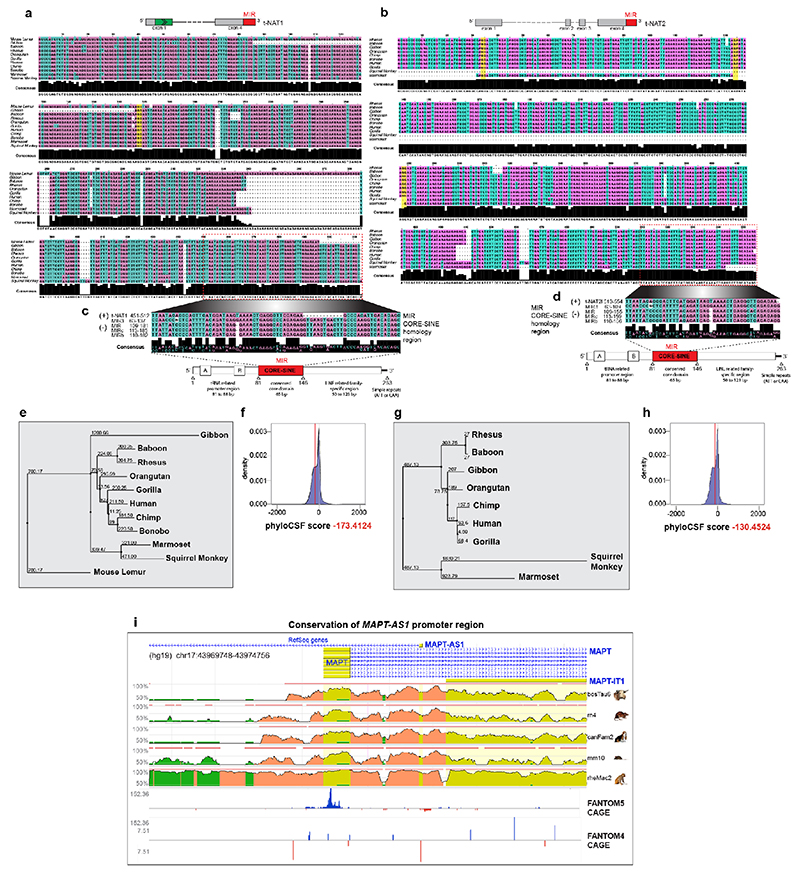
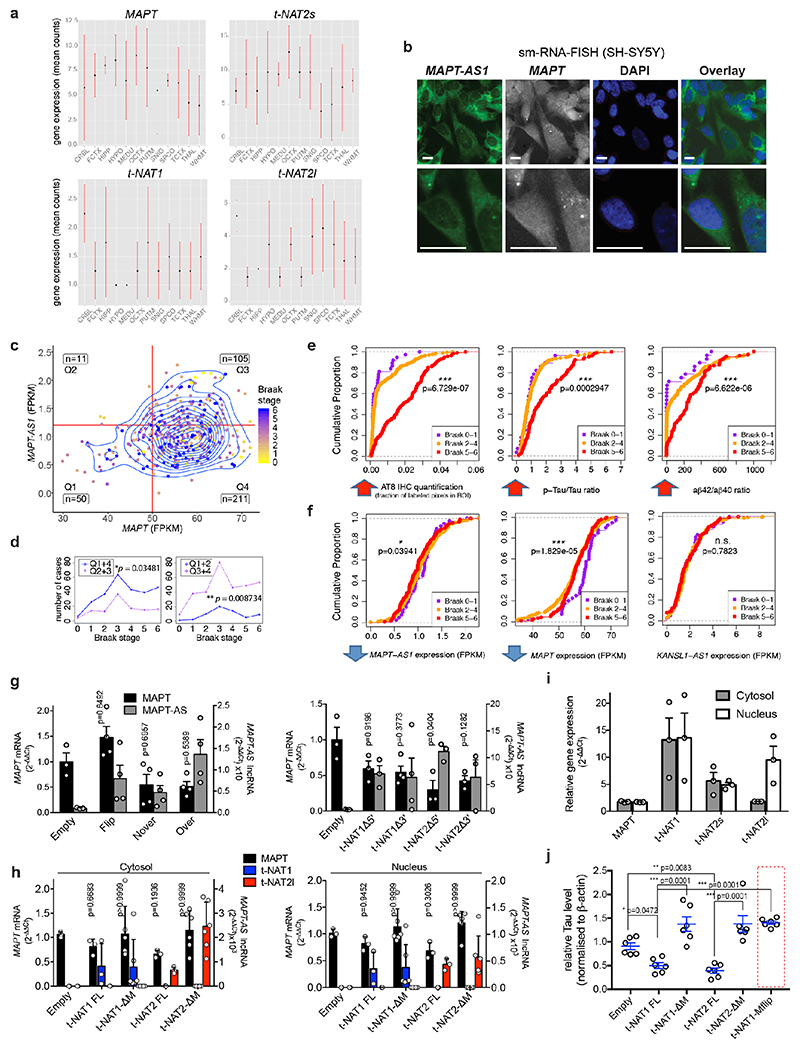
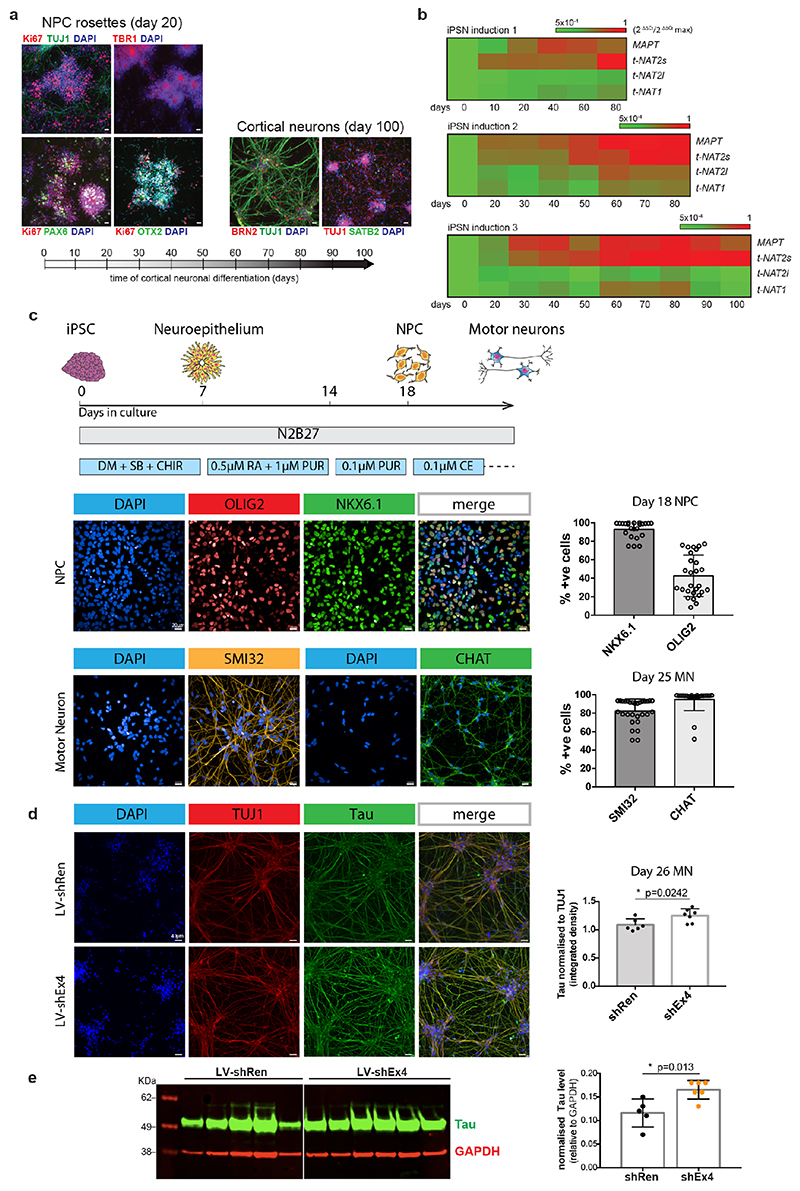
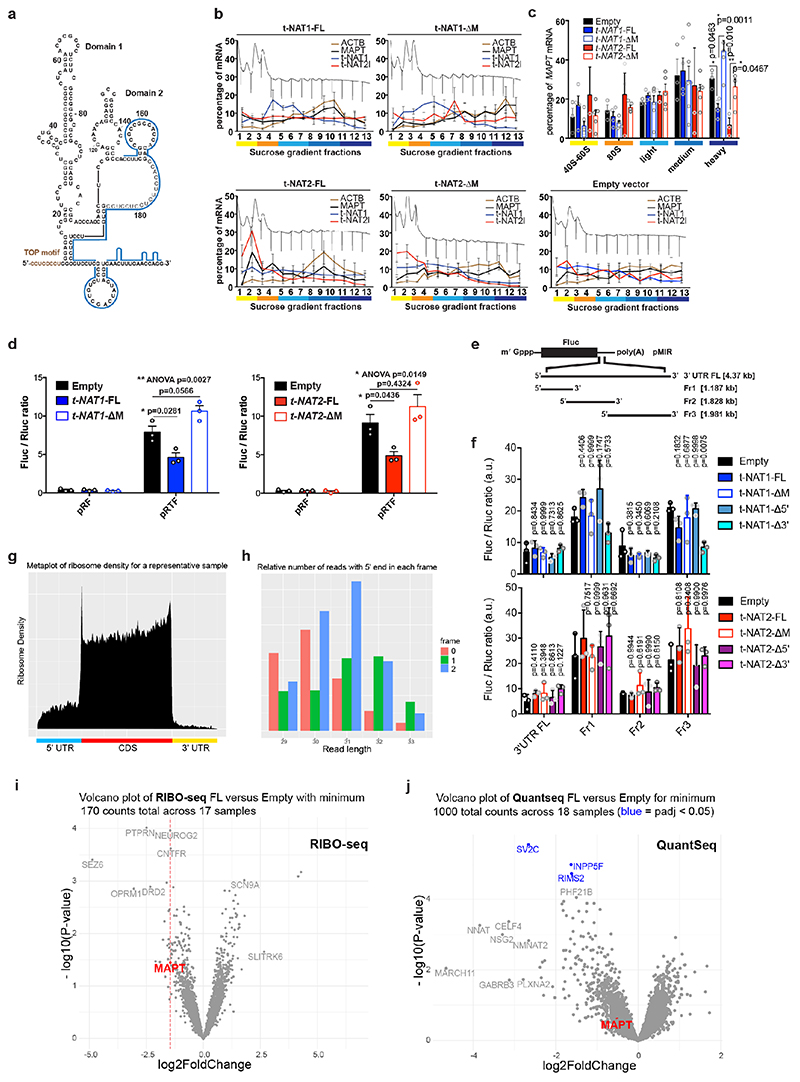
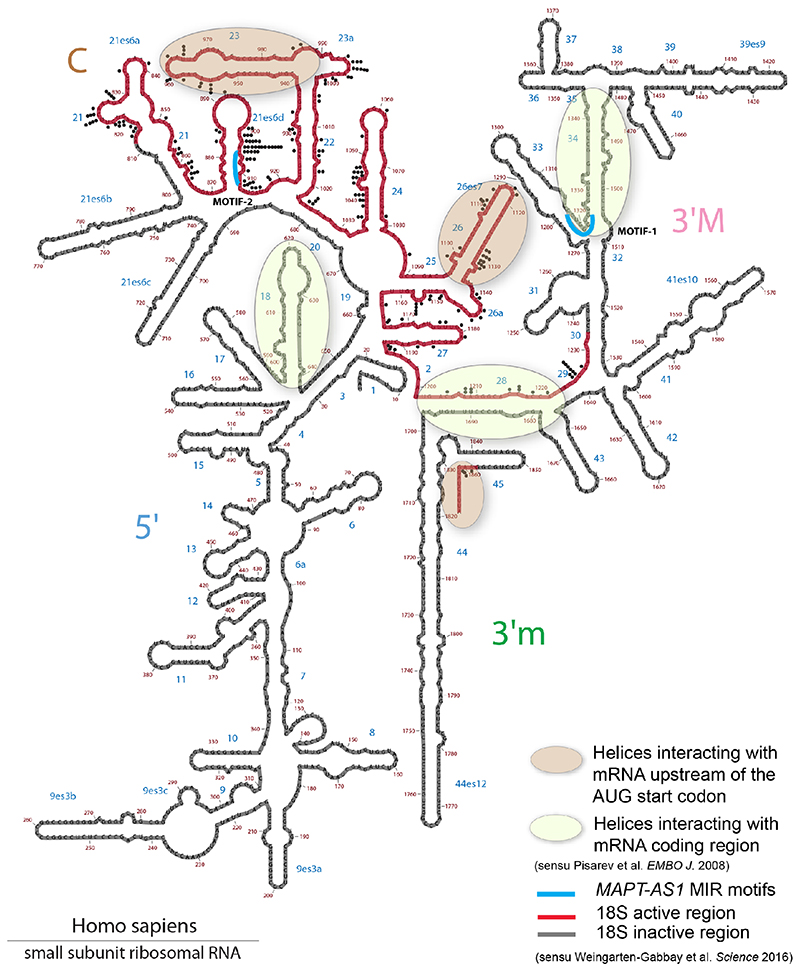
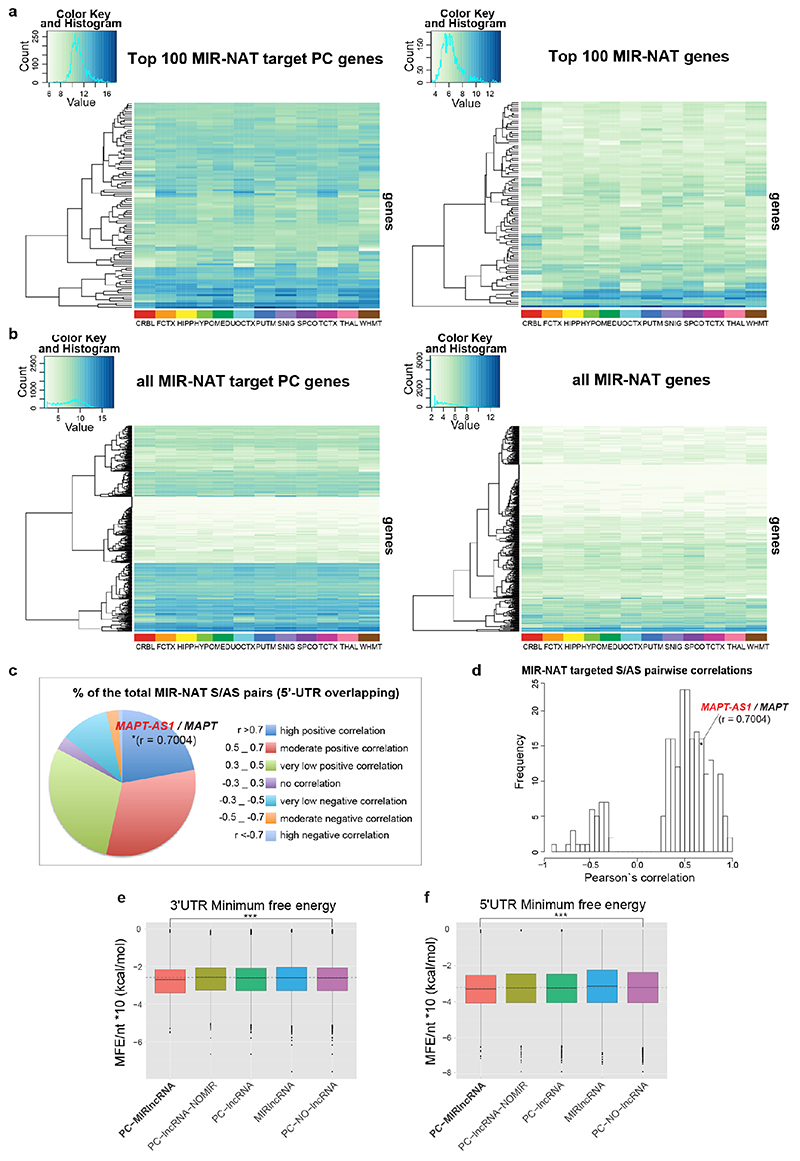
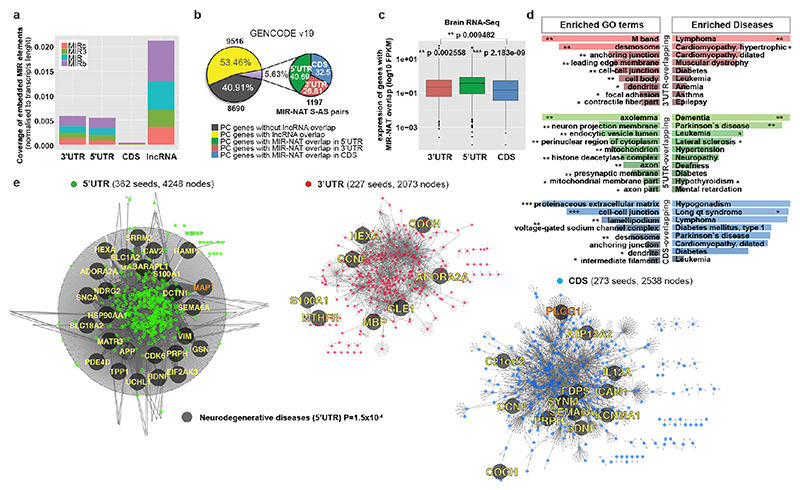
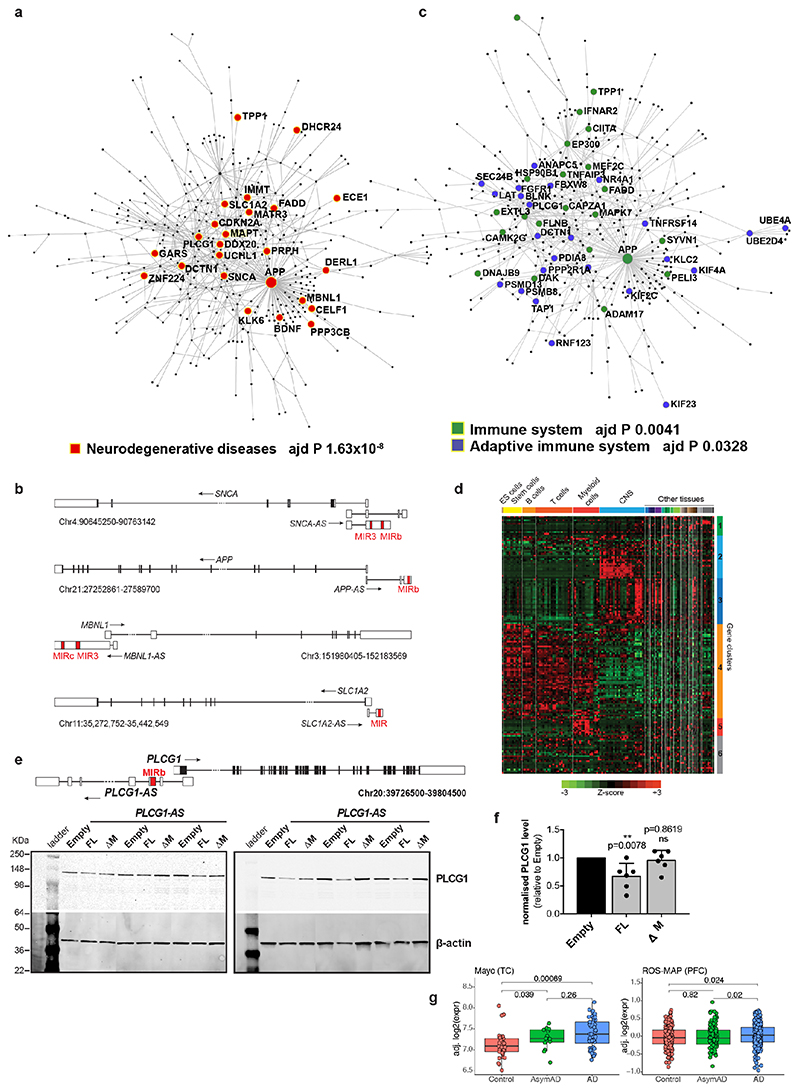
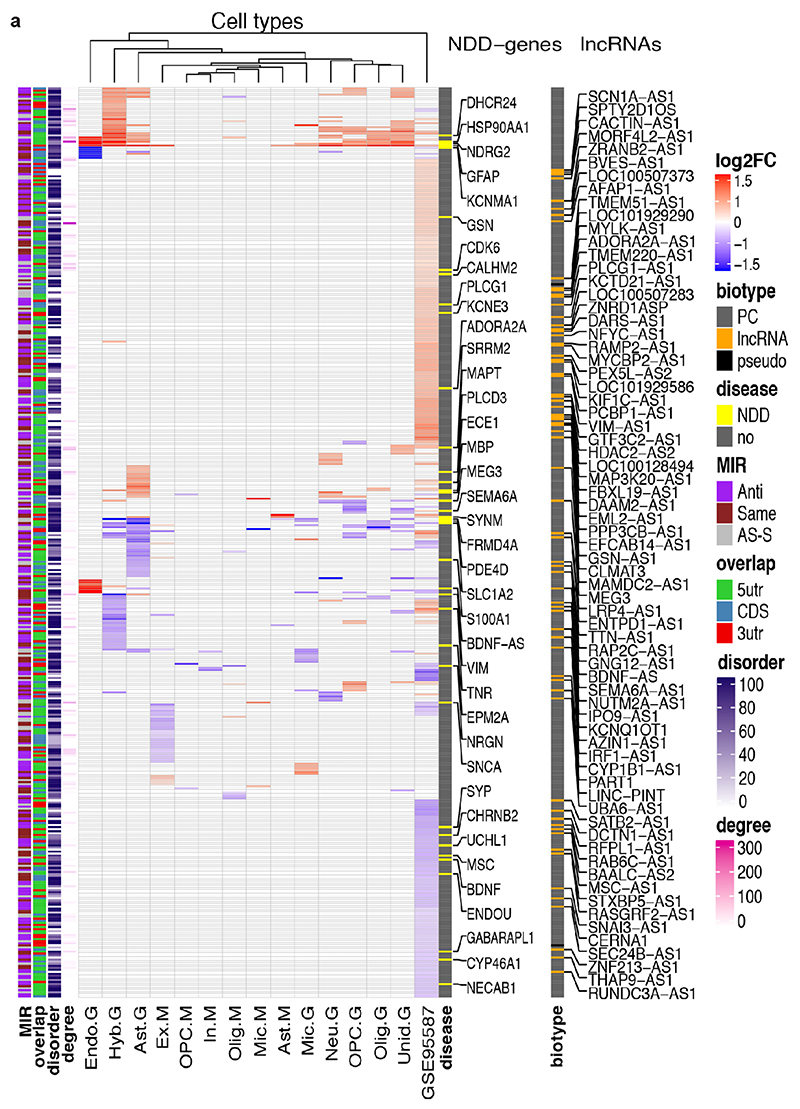
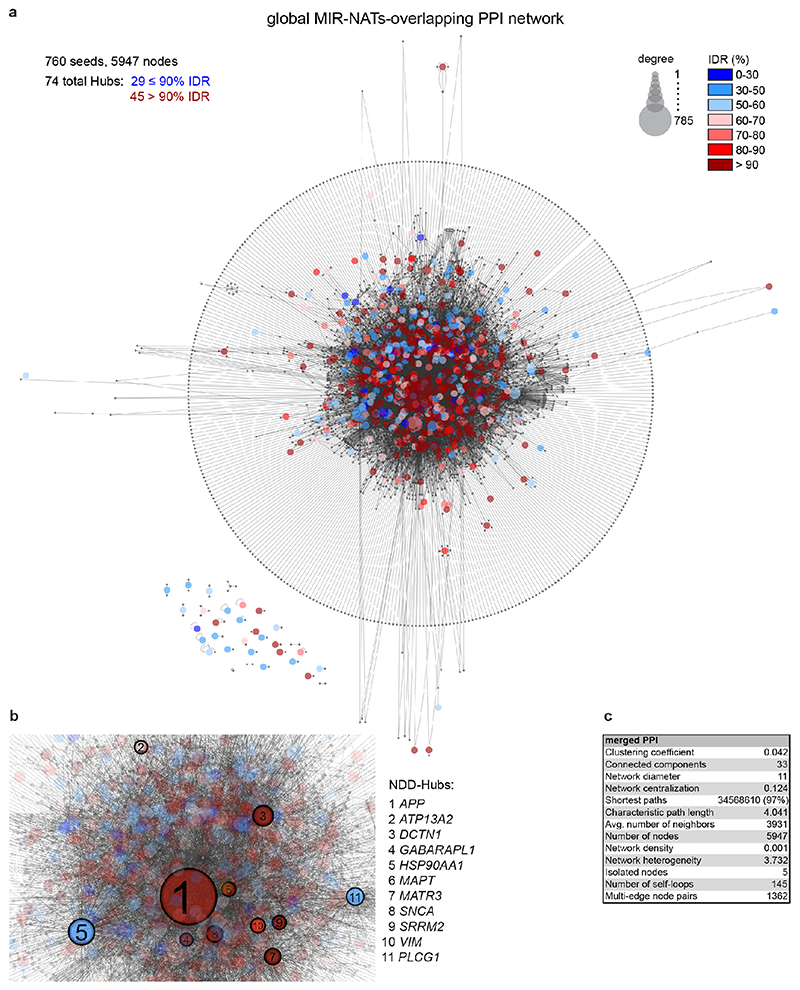
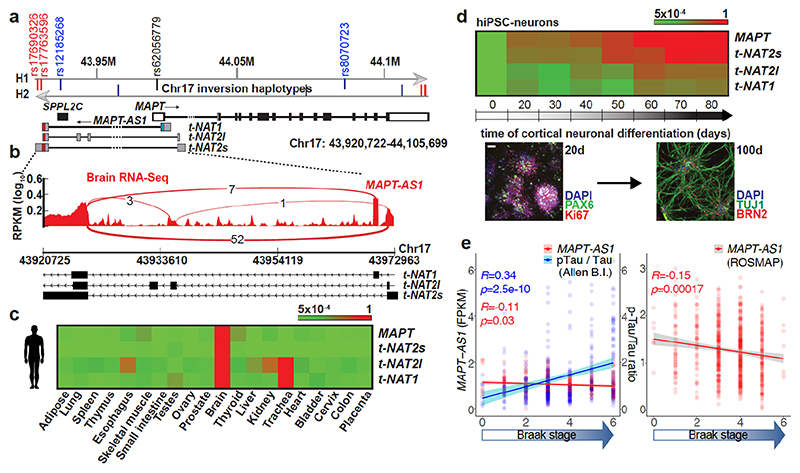
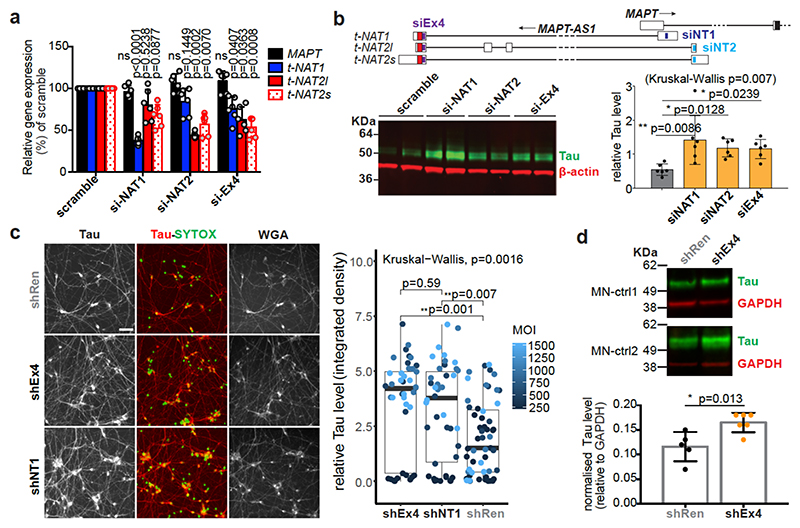
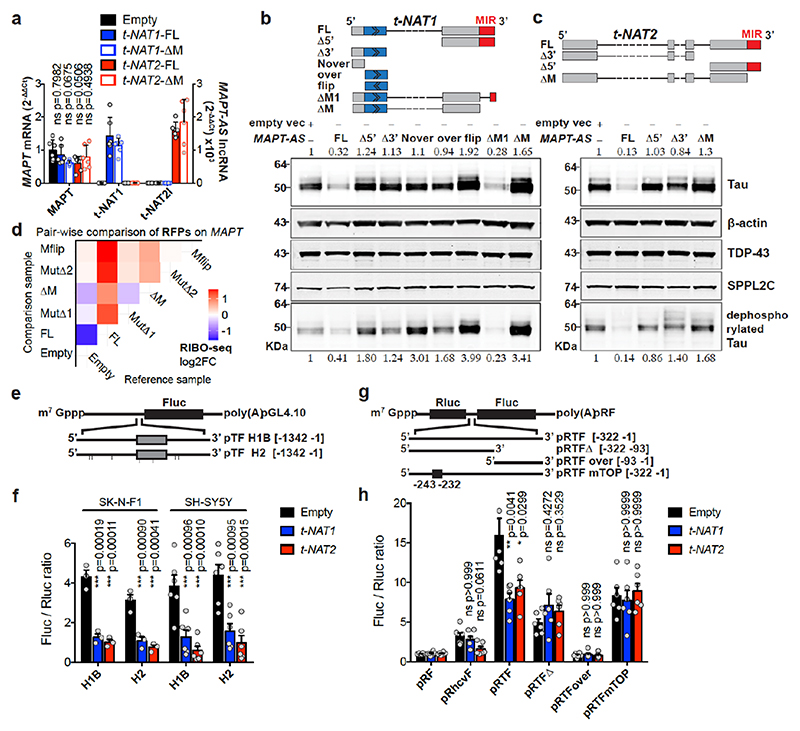
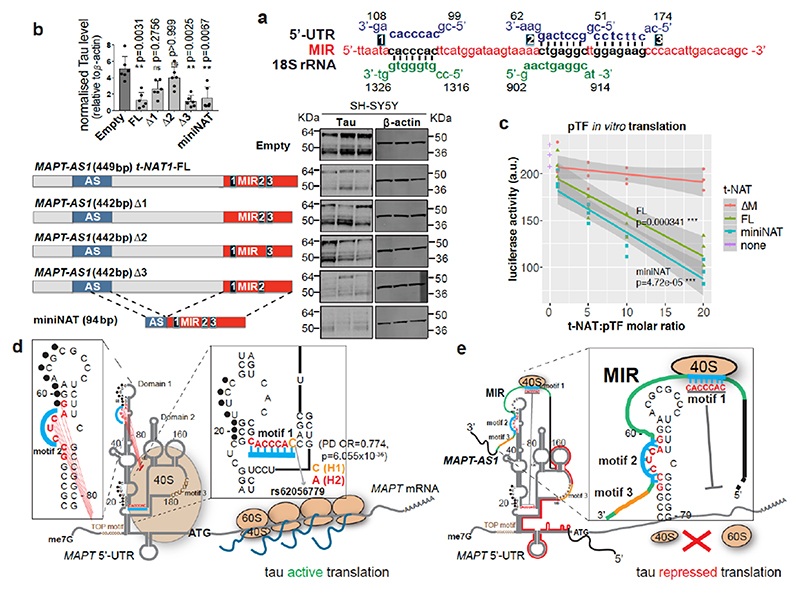
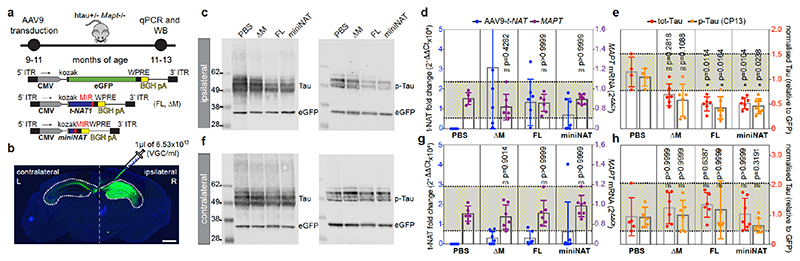
The human genome expresses thousands of natural antisense transcripts (NAT) that can regulate epigenetic state, transcription, RNA stability or translation of their overlapping genes1,2. Here we describe MAPT-AS1, a brain-enriched NAT that is conserved in primates and contains an embedded mammalian-wide interspersed repeat (MIR), which represses tau translation by competing for ribosomal RNA pairing with the MAPT mRNA internal ribosome entry site3. MAPT encodes tau, a neuronal intrinsically disordered protein (IDP) that stabilizes axonal microtubules. Hyperphosphorylated, aggregation-prone tau forms the hallmark inclusions of tauopathies4. Mutations in MAPT cause familial frontotemporal dementia, and common variations forming the MAPT H1 haplotype are a significant risk factor in many tauopathies5 and Parkinson's disease. Notably, expression of MAPT-AS1 or minimal essential sequences from MAPT-AS1 (including MIR) reduces-whereas silencing MAPT-AS1 expression increases-neuronal tau levels, and correlate with tau pathology in human brain. Moreover, we identified many additional NATs with embedded MIRs (MIR-NATs), which are overrepresented at coding genes linked to neurodegeneration and/or encoding IDPs, and confirmed MIR-NAT-mediated translational control of one such gene, PLCG1. These results demonstrate a key role for MAPT-AS1 in tauopathies and reveal a potentially broad contribution of MIR-NATs to the tightly controlled translation of IDPs6, with particular relevance for proteostasis in neurodegeneration.
Conflict of interest statement
The authors (R.S. and R.dS.) declare the following competing interest: Patent WO2017199041A1
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
- FC001002/CRUK_/Cancer Research UK/United Kingdom
- G1001253/MRC_/Medical Research Council/United Kingdom
- G-1307/PUK_/Parkinson's UK/United Kingdom
- 202903/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- MR/N026004/1/MRC_/Medical Research Council/United Kingdom
- MR/L023784/2/MRC_/Medical Research Council/United Kingdom
- MR/L501542/1/MRC_/Medical Research Council/United Kingdom
- G0501560/MRC_/Medical Research Council/United Kingdom
- MR/K01417X/1/MRC_/Medical Research Council/United Kingdom
- G-0907/PUK_/Parkinson's UK/United Kingdom
- G0701075/MRC_/Medical Research Council/United Kingdom
- MR/S006591/1/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- K-1212/PUK_/Parkinson's UK/United Kingdom
- MR/N008324/1/MRC_/Medical Research Council/United Kingdom
- G0901254/MRC_/Medical Research Council/United Kingdom
- MR/M02492X/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
