

Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia
- PMID: 34012921
- PMCID: PMC8126717
- DOI: 10.3389/fonc.2021.659253
Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia
Abstract
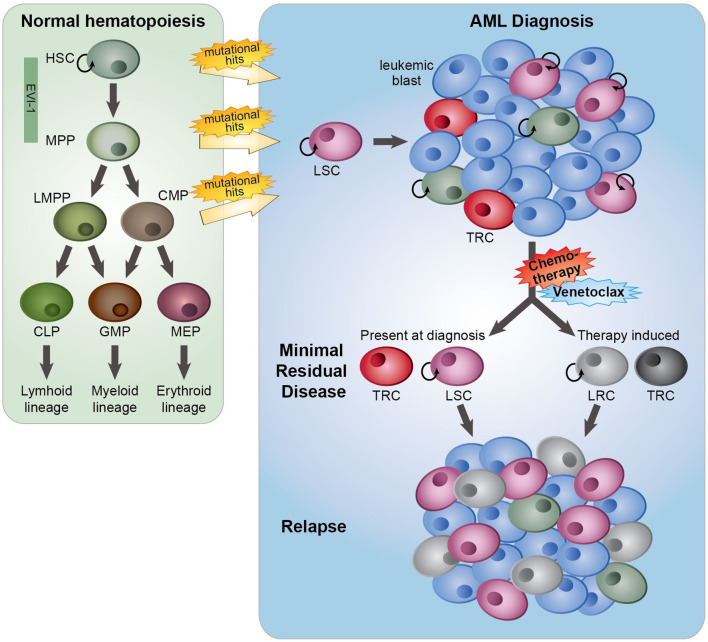
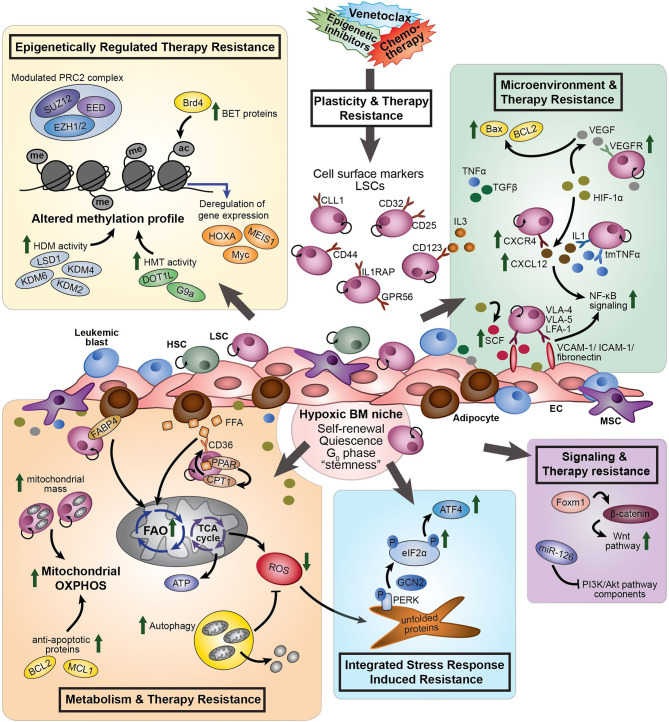
Standard induction chemotherapy, consisting of an anthracycline and cytarabine, has been the first-line therapy for many years to treat acute myeloid leukemia (AML). Although this treatment induces complete remissions in the majority of patients, many face a relapse (adaptive resistance) or have refractory disease (primary resistance). Moreover, older patients are often unfit for cytotoxic-based treatment. AML relapse is due to the survival of therapy-resistant leukemia cells (minimal residual disease, MRD). Leukemia cells with stem cell features, named leukemic stem cells (LSCs), residing within MRD are thought to be at the origin of relapse initiation. It is increasingly recognized that leukemia "persisters" are caused by intra-leukemic heterogeneity and non-genetic factors leading to plasticity in therapy response. The BCL2 inhibitor venetoclax, combined with hypomethylating agents or low dose cytarabine, represents an important new therapy especially for older AML patients. However, often there is also a small population of AML cells refractory to venetoclax treatment. As AML MRD reflects the sum of therapy resistance mechanisms, the different faces of treatment "persisters" and LSCs might be exploited to reach an optimal therapy response and prevent the initiation of relapse. Here, we describe the different epigenetic, transcriptional, and metabolic states of therapy sensitive and resistant AML (stem) cell populations and LSCs, how these cell states are influenced by the microenvironment and affect treatment outcome of AML. Moreover, we discuss potential strategies to target dynamic treatment resistance and LSCs.
Keywords: acute myeloid leukemia; leukemic stem cells; minimal residual disease; plasticity; therapy resistance.
Copyright © 2021 van Gils, Denkers and Smit.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Terwijn M, van Putten WLJ, Kelder A, van der Velden VHJ, Brooimans RA, Pabst T, et al. . High Prognostic Impact of Flow Cytometric Minimal Residual Disease Detection in Acute Myeloid Leukemia: Data From the HOVON/SAKK Aml 42a Study. J Clin Oncol (2013) 31:3889–97. 10.1200/JCO.2012.45.9628 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials