

Molecular clock REV-ERBα regulates cigarette smoke-induced pulmonary inflammation and epithelial-mesenchymal transition
- PMID: 34014841
- PMCID: PMC8262497
- DOI: 10.1172/jci.insight.145200
Molecular clock REV-ERBα regulates cigarette smoke-induced pulmonary inflammation and epithelial-mesenchymal transition
Abstract
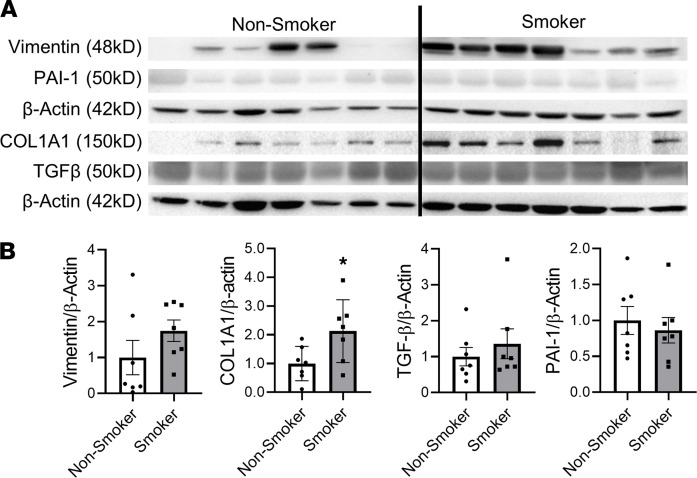
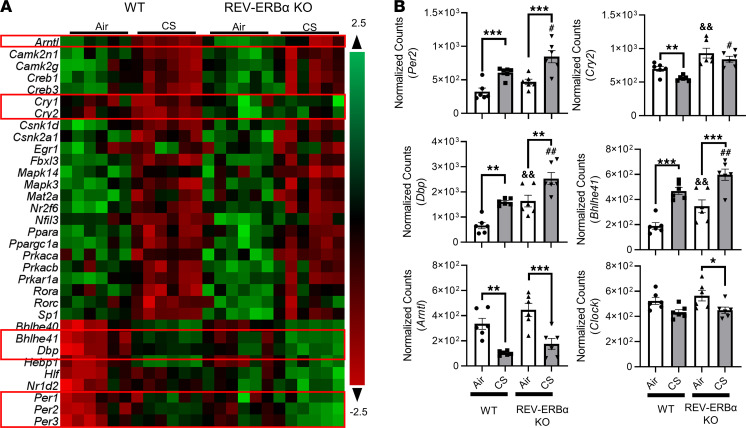
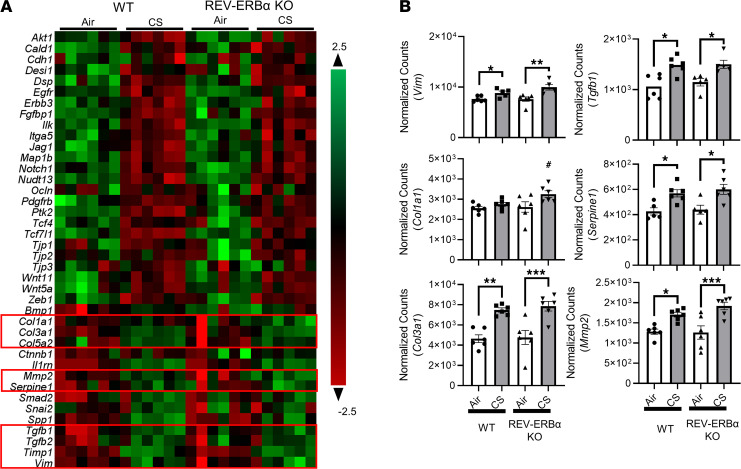
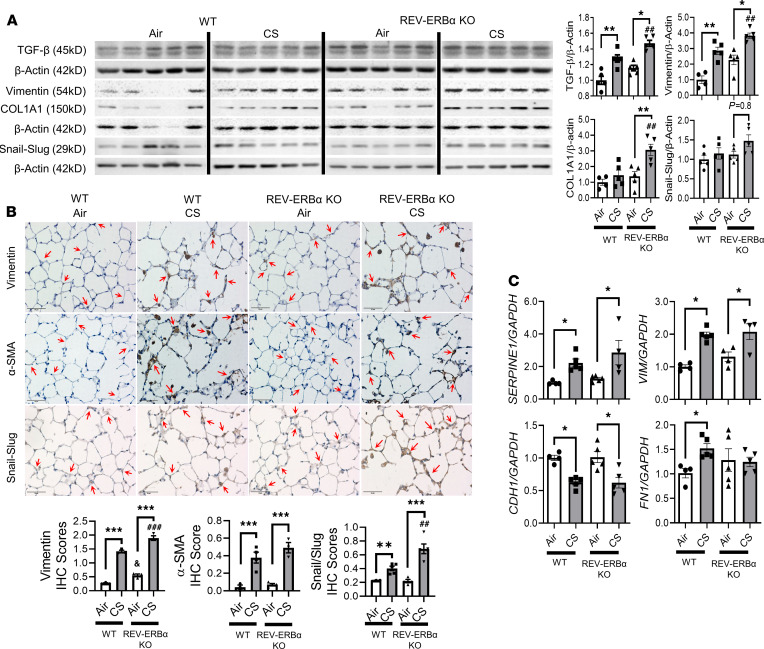
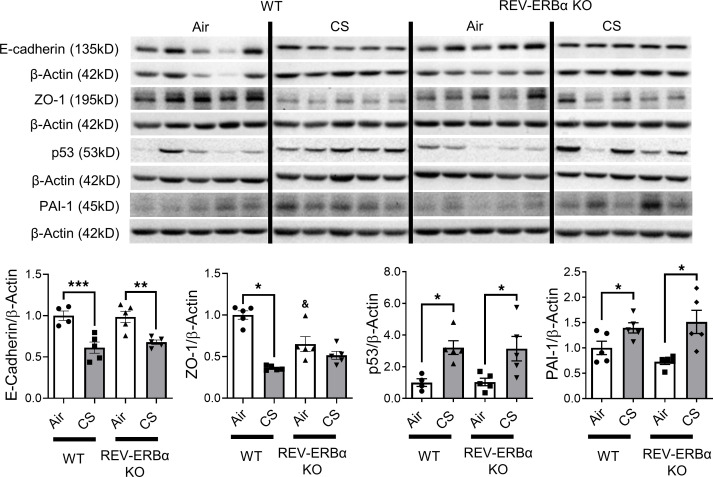
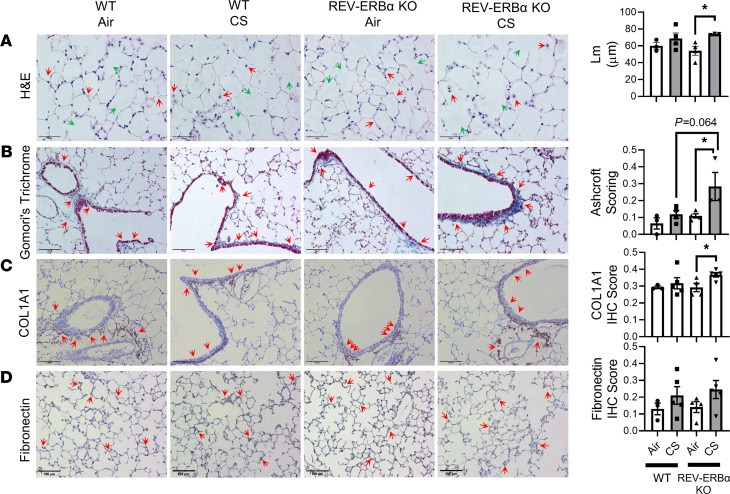
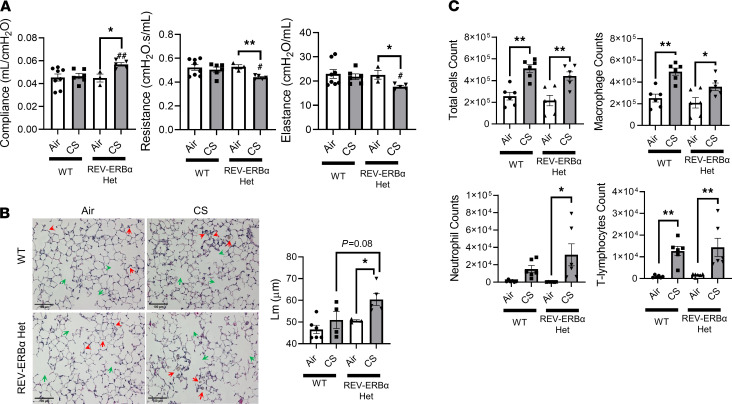
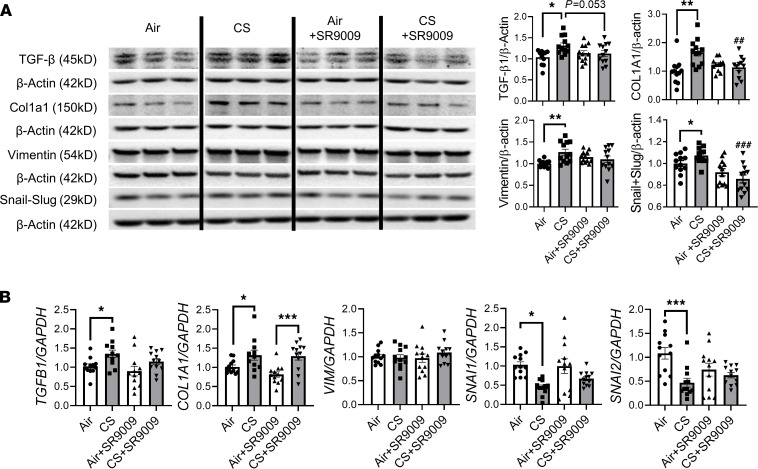
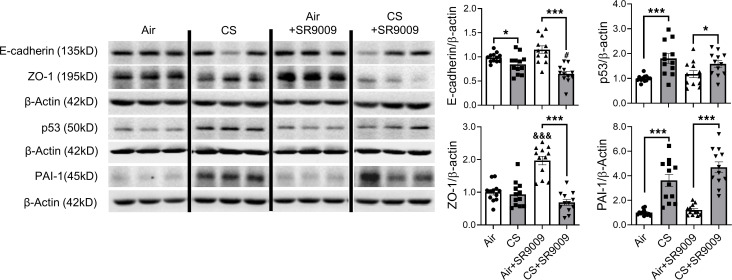
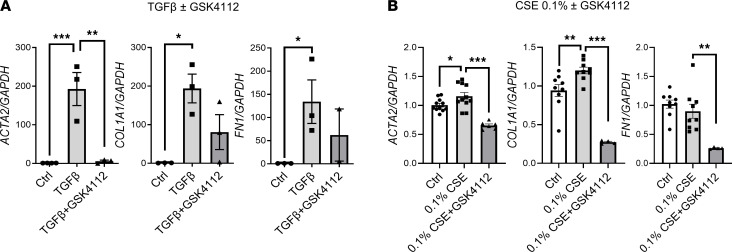
Cigarette smoke (CS) is the main etiological factor in the pathogenesis of emphysema/chronic obstructive pulmonary disease (COPD), which is associated with abnormal epithelial-mesenchymal transition (EMT). Previously, we have shown an association among circadian rhythms, CS-induced lung inflammation, and nuclear heme receptor α (REV-ERBα), acting as an antiinflammatory target in both pulmonary epithelial cells and fibroblasts. We hypothesized that molecular clock REV-ERBα plays an important role in CS-induced circadian dysfunction and EMT alteration. C57BL/6J WT and REV-ERBα heterozygous (Het) and -KO mice were exposed to CS for 30 days (subchronic) and 4 months (chronic), and WT mice were exposed to CS for 10 days with or without REV-ERBα agonist (SR9009) administration. Subchronic/chronic CS exposure caused circadian disruption and dysregulated EMT in the lungs of WT and REV-ERBα-KO mice; both circadian and EMT dysregulation were exaggerated in the REV-ERBα-KO condition. REV-ERBα agonist, SR9009 treatment reduced acute CS-induced inflammatory response and abnormal EMT in the lungs. Moreover, REV-ERBα agonist (GSK4112) inhibited TGF-β/CS-induced fibroblast differentiation in human fetal lung fibroblast 1 (HFL-1). Thus, CS-induced circadian gene alterations and EMT activation are mediated through a Rev-erbα-dependent mechanism, which suggests activation of REV-ERBα as a novel therapeutic approach for smoking-induced chronic inflammatory lung diseases.
Keywords: COPD; Inflammation; Pulmonology.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
