

Very long intergenic non-coding (vlinc) RNAs directly regulate multiple genes in cis and trans
- PMID: 34016118
- PMCID: PMC8139166
- DOI: 10.1186/s12915-021-01044-x
Very long intergenic non-coding (vlinc) RNAs directly regulate multiple genes in cis and trans
Abstract
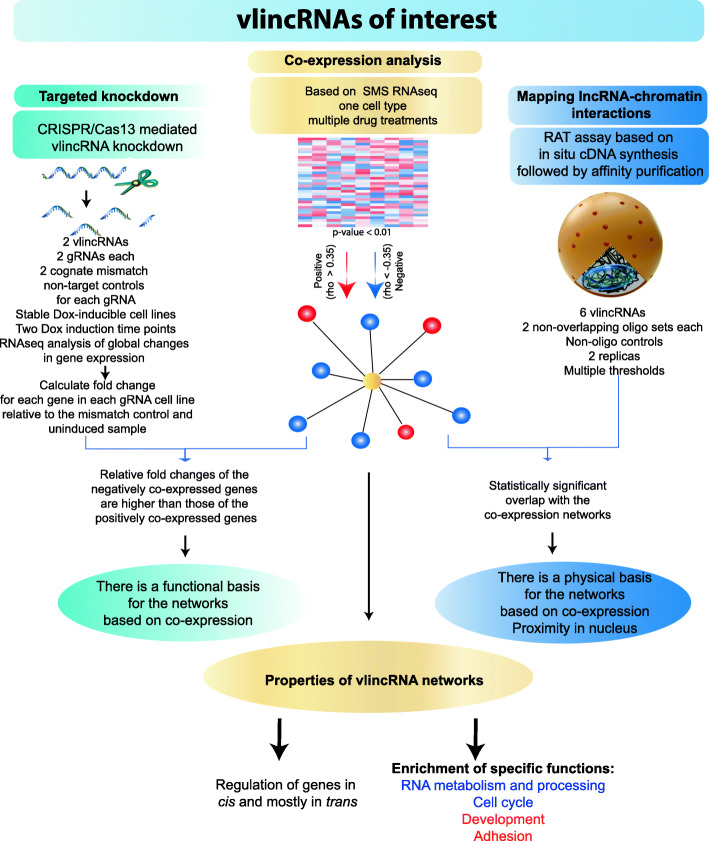
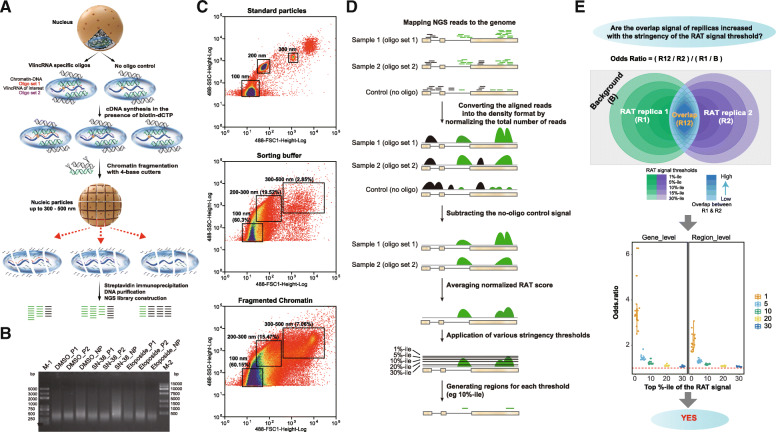
Background: The majority of the human genome is transcribed in the form of long non-coding (lnc) RNAs. While these transcripts have attracted considerable interest, their molecular mechanisms of function and biological significance remain controversial. One of the main reasons behind this lies in the significant challenges posed by lncRNAs requiring the development of novel methods and concepts to unravel their functionality. Existing methods often lack cross-validation and independent confirmation by different methodologies and therefore leave significant ambiguity as to the authenticity of the outcomes. Nonetheless, despite all the caveats, it appears that lncRNAs may function, at least in part, by regulating other genes via chromatin interactions. Therefore, the function of a lncRNA could be inferred from the function of genes it regulates. In this work, we present a genome-wide functional annotation strategy for lncRNAs based on identification of their regulatory networks via the integration of three distinct types of approaches: co-expression analysis, mapping of lncRNA-chromatin interactions, and assaying molecular effects of lncRNA knockdowns obtained using an inducible and highly specific CRISPR/Cas13 system.
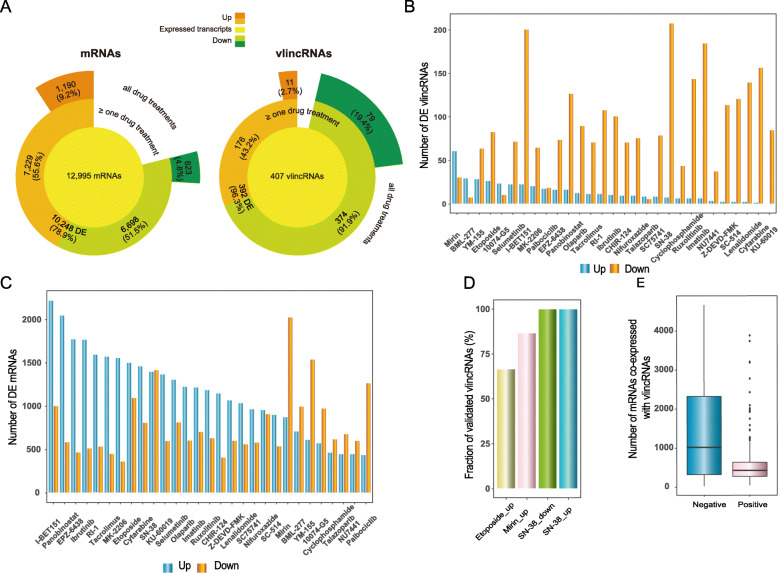
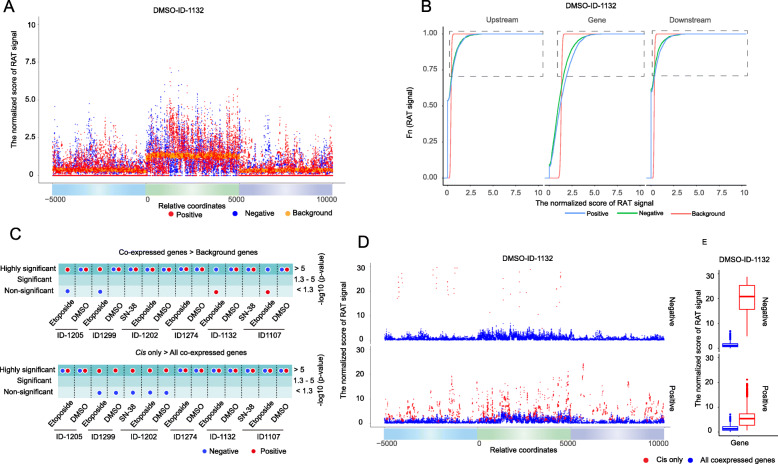
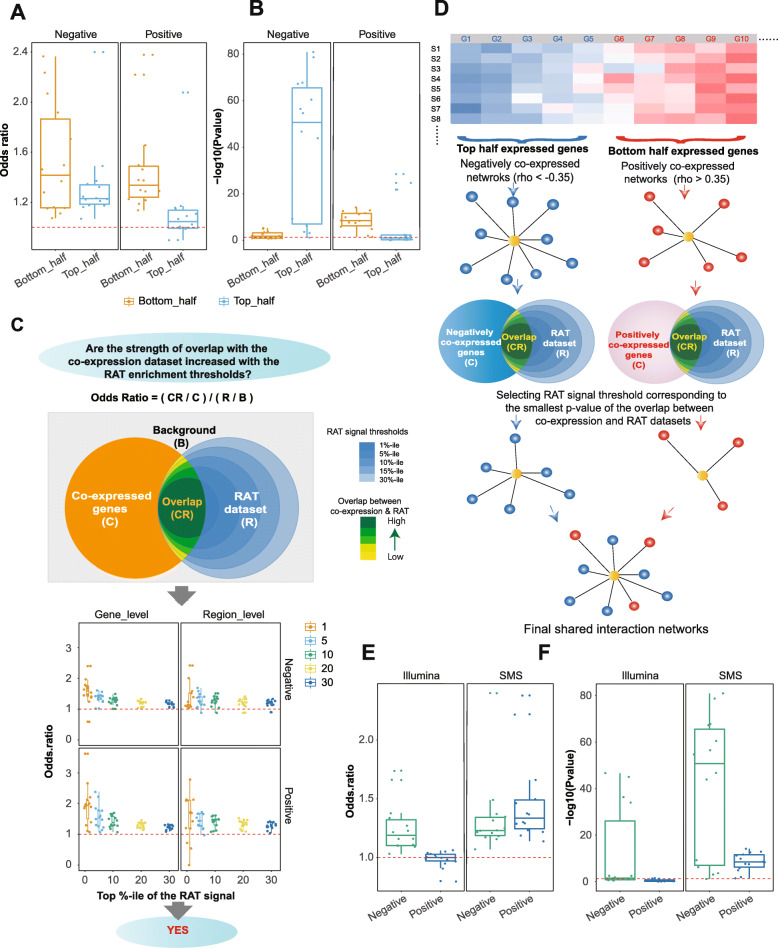
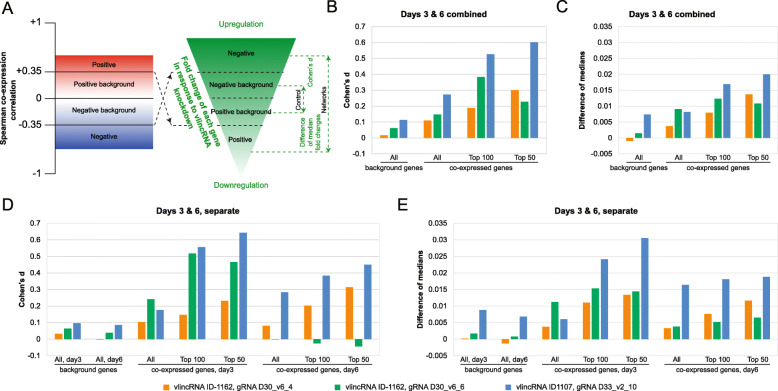
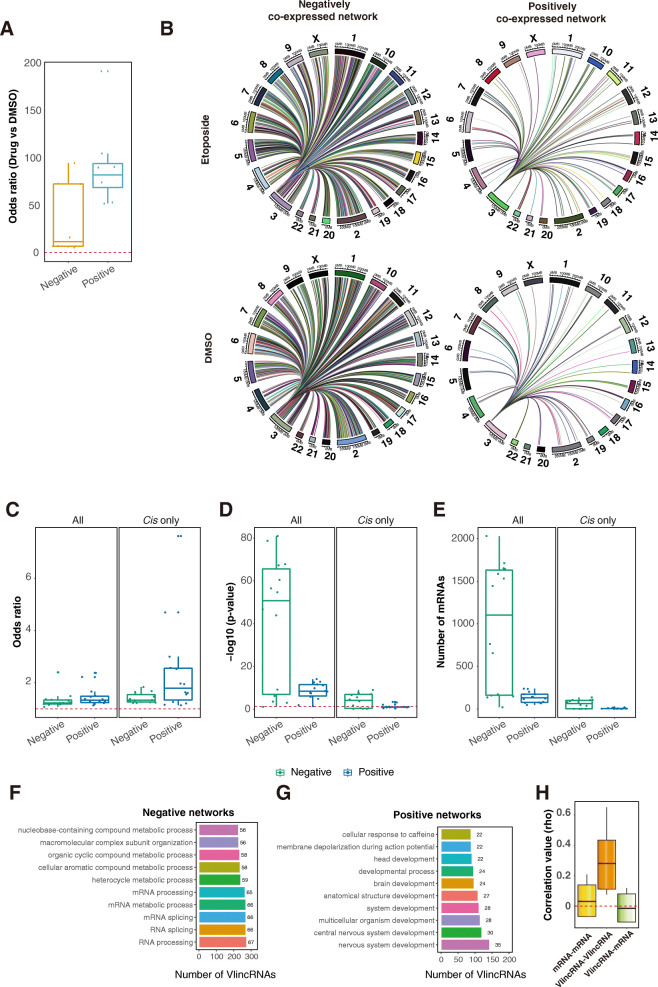
Results: We applied the strategy to annotate 407 very long intergenic non-coding (vlinc) RNAs belonging to a novel widespread subclass of lncRNAs. We show that vlincRNAs indeed appear to regulate multiple genes encoding proteins predominantly involved in RNA- and development-related functions, cell cycle, and cellular adhesion via a mechanism involving proximity between vlincRNAs and their targets in the nucleus. A typical vlincRNAs can be both a positive and negative regulator and regulate multiple genes both in trans and cis. Finally, we show vlincRNAs and their regulatory networks potentially represent novel components of DNA damage response and are functionally important for the ability of cancer cells to survive genotoxic stress.
Conclusions: This study provides strong evidence for the regulatory role of the vlincRNA class of lncRNAs and a potentially important role played by these transcripts in the hidden layer of RNA-based regulation in complex biological systems.
Keywords: Anti-cancer drugs; CRISPR/Cas13; Cell cycle; Development; RNA processing; RNA-chromatin interactions; Regulatory networks; Single-molecule sequencing; lncRNA; vlincRNA.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response.Sci Rep. 2020 Feb 4;10(1):1794. doi: 10.1038/s41598-020-58104-5. Sci Rep. 2020. PMID: 32020014 Free PMC article.
-
Direct Interactions with Nascent Transcripts Is Potentially a Common Targeting Mechanism of Long Non-Coding RNAs.Genes (Basel). 2020 Dec 10;11(12):1483. doi: 10.3390/genes11121483. Genes (Basel). 2020. PMID: 33321875 Free PMC article.
-
Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress.BMC Plant Biol. 2018 Jan 25;18(1):23. doi: 10.1186/s12870-018-1238-0. BMC Plant Biol. 2018. PMID: 29370759 Free PMC article.
-
The Properties of Long Noncoding RNAs That Regulate Chromatin.Annu Rev Genomics Hum Genet. 2016 Aug 31;17:69-94. doi: 10.1146/annurev-genom-090314-024939. Epub 2016 Apr 21. Annu Rev Genomics Hum Genet. 2016. PMID: 27147088 Review.
-
Biological Function of Long Non-coding RNA (LncRNA) Xist.Front Cell Dev Biol. 2021 Jun 10;9:645647. doi: 10.3389/fcell.2021.645647. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34178980 Free PMC article. Review.
Cited by
-
LncRNA Osilr9 coordinates promoter DNA demethylation and the intrachromosomal loop structure required for maintaining stem cell pluripotency.Mol Ther. 2023 Jun 7;31(6):1791-1806. doi: 10.1016/j.ymthe.2022.12.010. Epub 2022 Dec 15. Mol Ther. 2023. PMID: 36523163 Free PMC article.
-
Chromatin lncRNA Platr10 controls stem cell pluripotency by coordinating an intrachromosomal regulatory network.Genome Biol. 2021 Aug 19;22(1):233. doi: 10.1186/s13059-021-02444-6. Genome Biol. 2021. PMID: 34412677 Free PMC article.
-
Endogenous retroelements in hematological malignancies: From epigenetic dysregulation to therapeutic targeting.Am J Hematol. 2025 Jan;100(1):116-130. doi: 10.1002/ajh.27501. Epub 2024 Oct 10. Am J Hematol. 2025. PMID: 39387681 Free PMC article. Review.
-
Long Intergenic Non-Coding RNAs of Human Chromosome 18: Focus on Cancers.Biomedicines. 2024 Feb 28;12(3):544. doi: 10.3390/biomedicines12030544. Biomedicines. 2024. PMID: 38540157 Free PMC article. Review.
-
Hotspots of single-strand DNA "breakome" are enriched at transcriptional start sites of genes.Front Mol Biosci. 2022 Aug 15;9:895795. doi: 10.3389/fmolb.2022.895795. eCollection 2022. Front Mol Biosci. 2022. PMID: 36046604 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
