

Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer
- PMID: 34016182
- PMCID: PMC8136103
- DOI: 10.1186/s13073-021-00895-x
Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer
Abstract
Background: Circulating tumor DNA (ctDNA) offers minimally invasive means to repeatedly interrogate tumor genomes, providing opportunities to monitor clonal dynamics induced by metastasis and therapeutic selective pressures. In metastatic cancers, ctDNA profiling allows for simultaneous analysis of both local and distant sites of recurrence. Despite the promise of ctDNA sampling, its utility in real-time genetic monitoring remains largely unexplored.
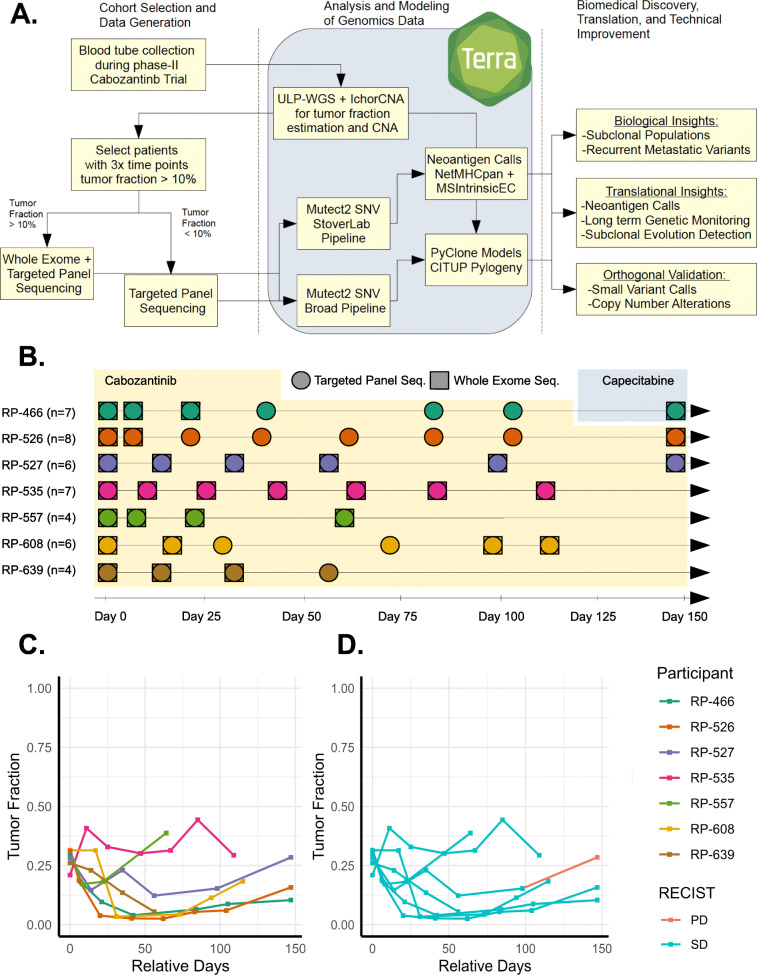
Methods: In this exploratory analysis, we characterize high-frequency ctDNA sample series collected over narrow time frames from seven patients with metastatic triple-negative breast cancer, each undergoing treatment with Cabozantinib, a multi-tyrosine kinase inhibitor (NCT01738438, https://clinicaltrials.gov/ct2/show/NCT01738438 ). Applying orthogonal whole exome sequencing, ultra-low pass whole genome sequencing, and 396-gene targeted panel sequencing, we analyzed 42 plasma-derived ctDNA libraries, representing 4-8 samples per patient with 6-42 days between samples. Integrating tumor fraction, copy number, and somatic variant information, we model tumor clonal dynamics, predict neoantigens, and evaluate consistency of genomic information from orthogonal assays.
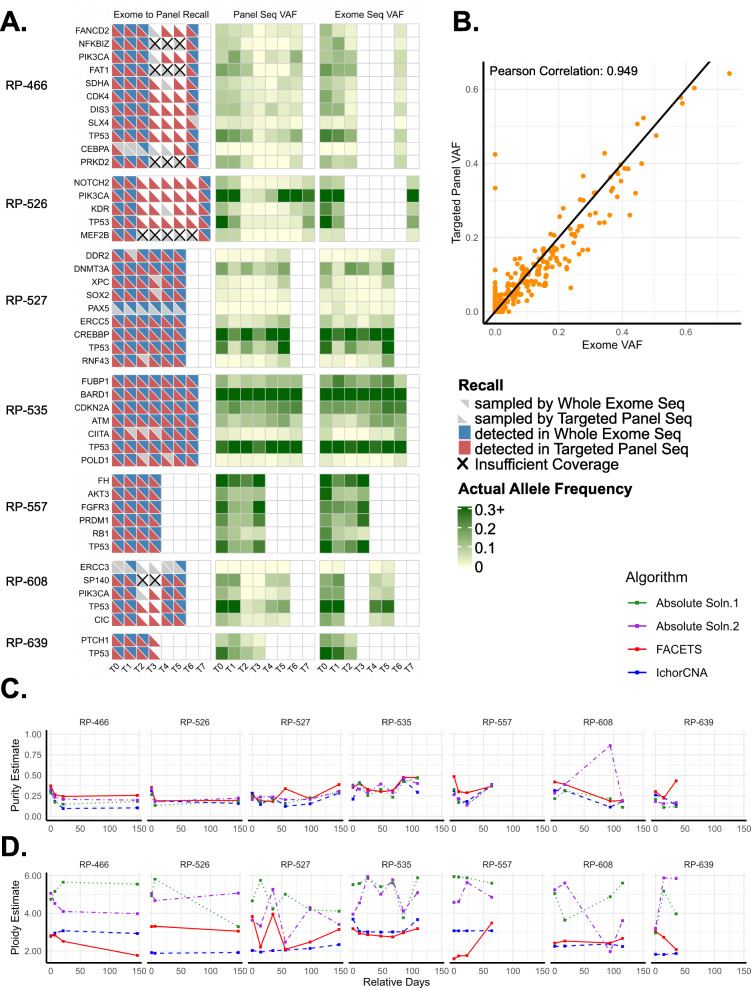
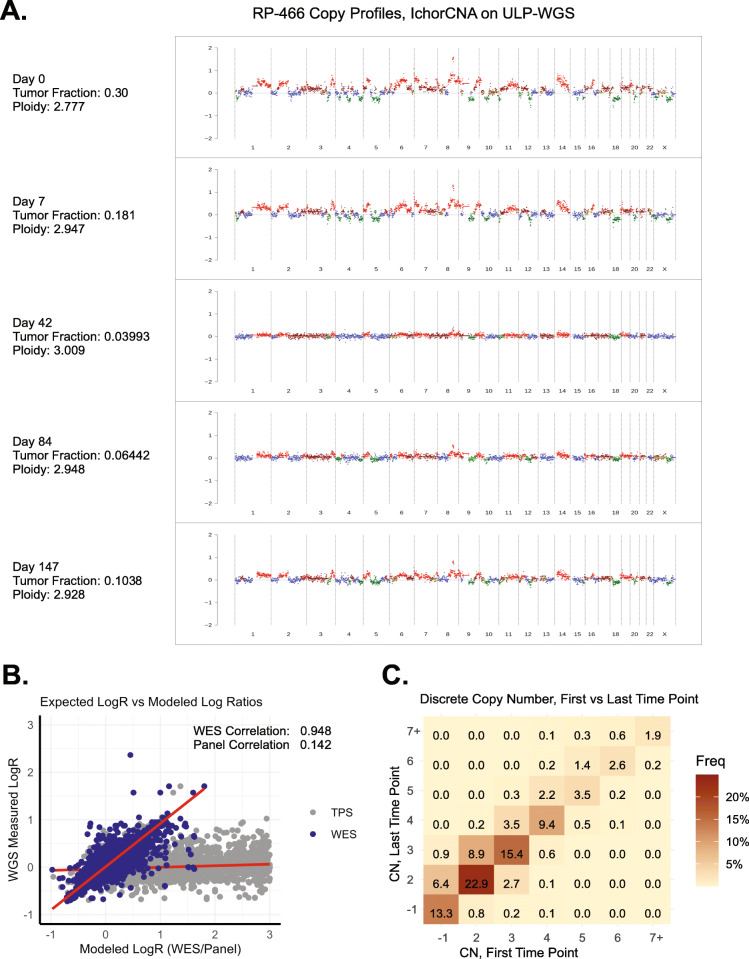
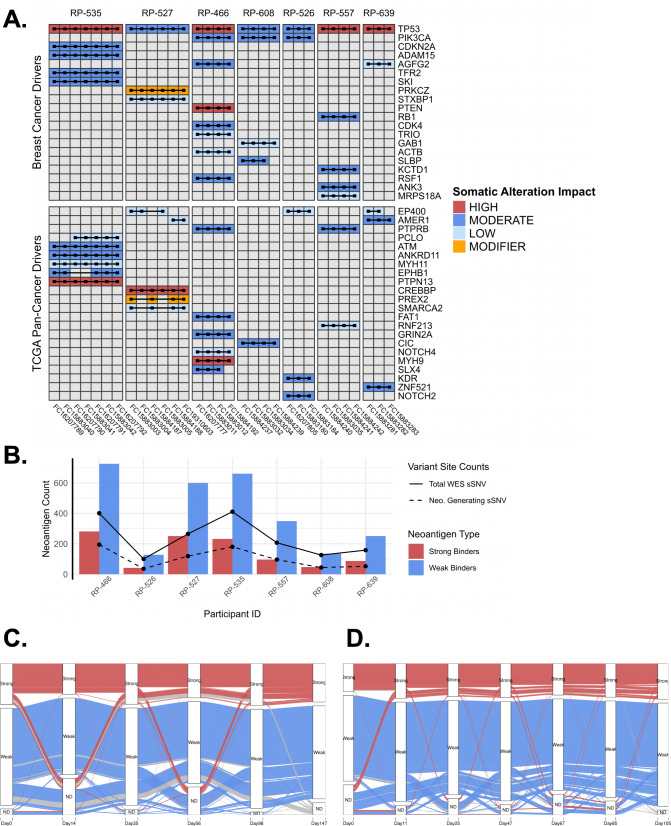
Results: We measured considerable variation in ctDNA tumor faction in each patient, often conflicting with RECIST imaging response metrics. In orthogonal sequencing, we found high concordance between targeted panel and whole exome sequencing in both variant detection and variant allele frequency estimation (specificity = 95.5%, VAF correlation, r = 0.949), Copy number remained generally stable, despite resolution limitations posed by low tumor fraction. Through modeling, we inferred and tracked distinct clonal populations specific to each patient and built phylogenetic trees revealing alterations in hallmark breast cancer drivers, including TP53, PIK3CA, CDK4, and PTEN. Our modeling revealed varied responses to therapy, with some individuals displaying stable clonal profiles, while others showed signs of substantial expansion or reduction in prevalence, with characteristic alterations of varied literature annotation in relation to the study drug. Finally, we predicted and tracked neoantigen-producing alterations across time, exposing translationally relevant detection patterns.
Conclusions: Despite technical challenges arising from low tumor content, metastatic ctDNA monitoring can aid our understanding of response and progression, while minimizing patient risk and discomfort. In this study, we demonstrate the potential for high-frequency monitoring of evolving genomic features, providing an important step toward scalable, translational genomics for clinical decision making.
Keywords: Circulating tumor DNA; Liquid biopsy; Neoantigens; Serial sequencing; Targeted panel sequencing; Tumor evolution; Ultra-low pass whole genome sequencing; ctDNA.
Conflict of interest statement
S.A.S. reported nonfinancial support from Bristol-Myers Squibb outside the submitted work. S.A.S. previously advised and has received consulting fees from Neon Therapeutics. S.A.S. reported nonfinancial support from Bristol-Myers Squibb, and equity in Agenus Inc., Agios Pharmaceuticals, Breakbio Corp., Bristol-Myers Squibb, Indiscine and Lumos Pharma, outside the submitted work. R.B.S. reported consulting Fees (e.g., advisory boards); Author; Roche, Merck, Eli Lilly. Fees for Non-CME Services Received Directly from Commercial Interest or their Agents (e.g., speakers’ bureaus); Author; Eli Lilly, Libbs, Novartis, Pfizer, ROCHE, Bristol-Myers Squibb. G. Ha: Receipt of Intellectual Property Rights/Patent Holder; Broad Institute. VAA reported advisory boards for AGCT GmbH and BerMs Inc. S.M.T. reported consulting fees (e.g., advisory boards); AstraZeneca, Lilly, Merck, Nektar, Novartis, Pfizer, Genentech/Roche, Immunomedics, Bristol-Myers Squibb, Eisai, Nanostring, Puma, Sanofi, Celldex, Odonate, Seattle Genetics, Daiichi Sankyo, Silverback Therapeutics, Abbvie, Athenex, OncoPep, Kyowa Kirin Pharmaceuticals, Samsung BioepsiVs. The remaining authors declare that they have no competing interests.
Figures

References
-
- Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61(4):1659–1665. - PubMed
-
- Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, Gydush G, Reed SC, Rotem D, Rhoades J, Loginov D, Livitz D, Rosebrock D, Leshchiner I, Kim J, Stewart C, Rosenberg M, Francis JM, Zhang CZ, Cohen O, Oh C, Ding H, Polak P, Lloyd M, Mahmud S, Helvie K, Merrill MS, Santiago RA, O’Connor EP, Jeong SH, Leeson R, Barry RM, Kramkowski JF, Zhang Z, Polacek L, Lohr JG, Schleicher M, Lipscomb E, Saltzman A, Oliver NM, Marini L, Waks AG, Harshman LC, Tolaney SM, van Allen EM, Winer EP, Lin NU, Nakabayashi M, Taplin ME, Johannessen CM, Garraway LA, Golub TR, Boehm JS, Wagle N, Getz G, Love JC, Meyerson M. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8(1):1324. doi: 10.1038/s41467-017-00965-y. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
