

Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the h ACE2 Receptor
- PMID: 34017819
- PMCID: PMC8129187
- DOI: 10.3389/fchem.2021.661230
Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the h ACE2 Receptor
Abstract
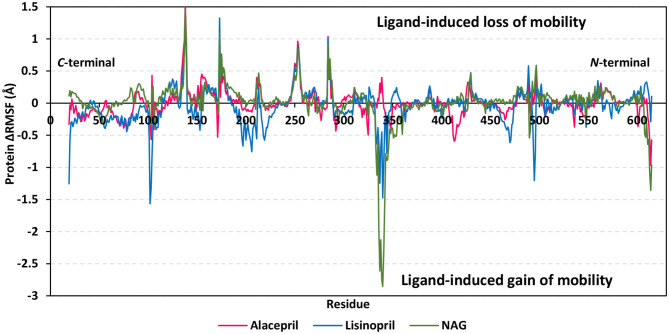
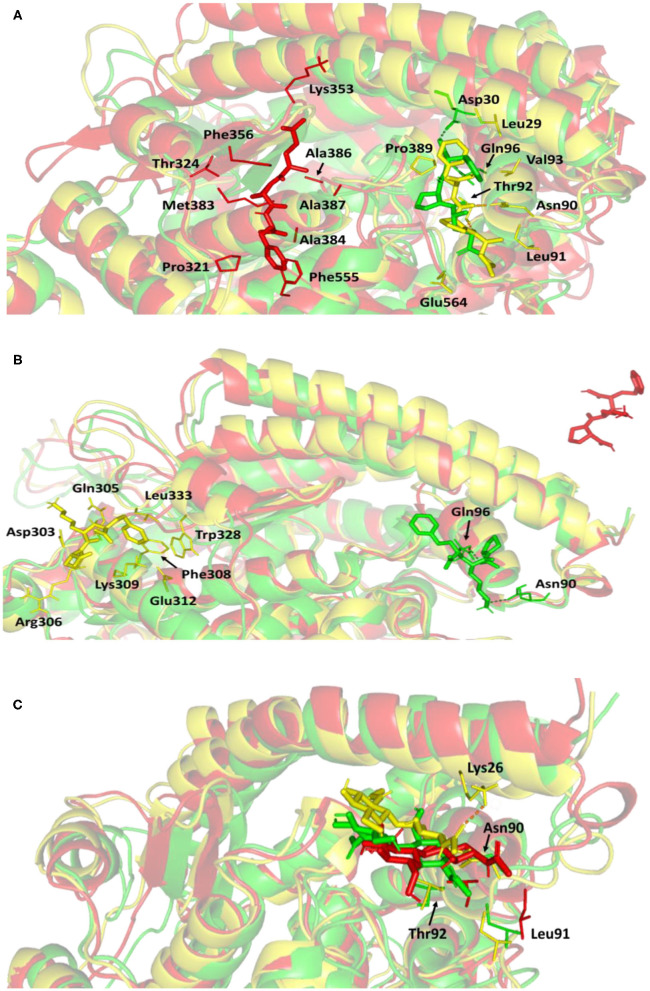
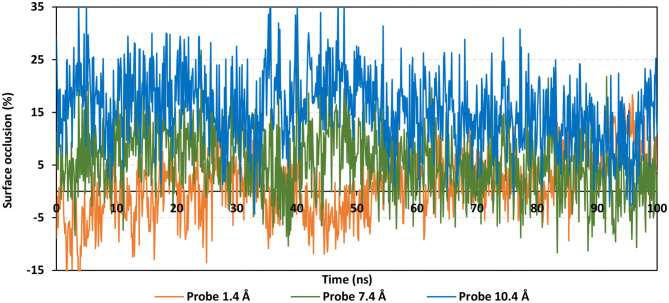
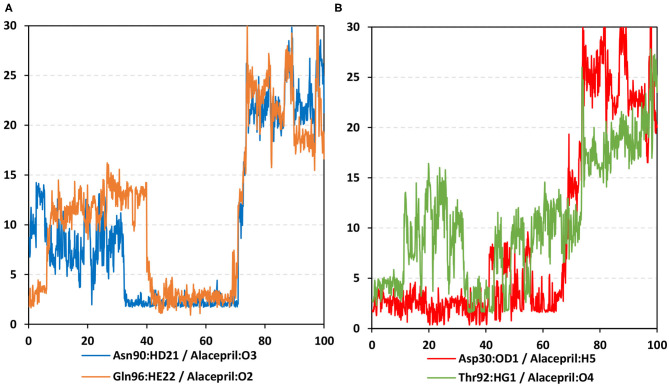
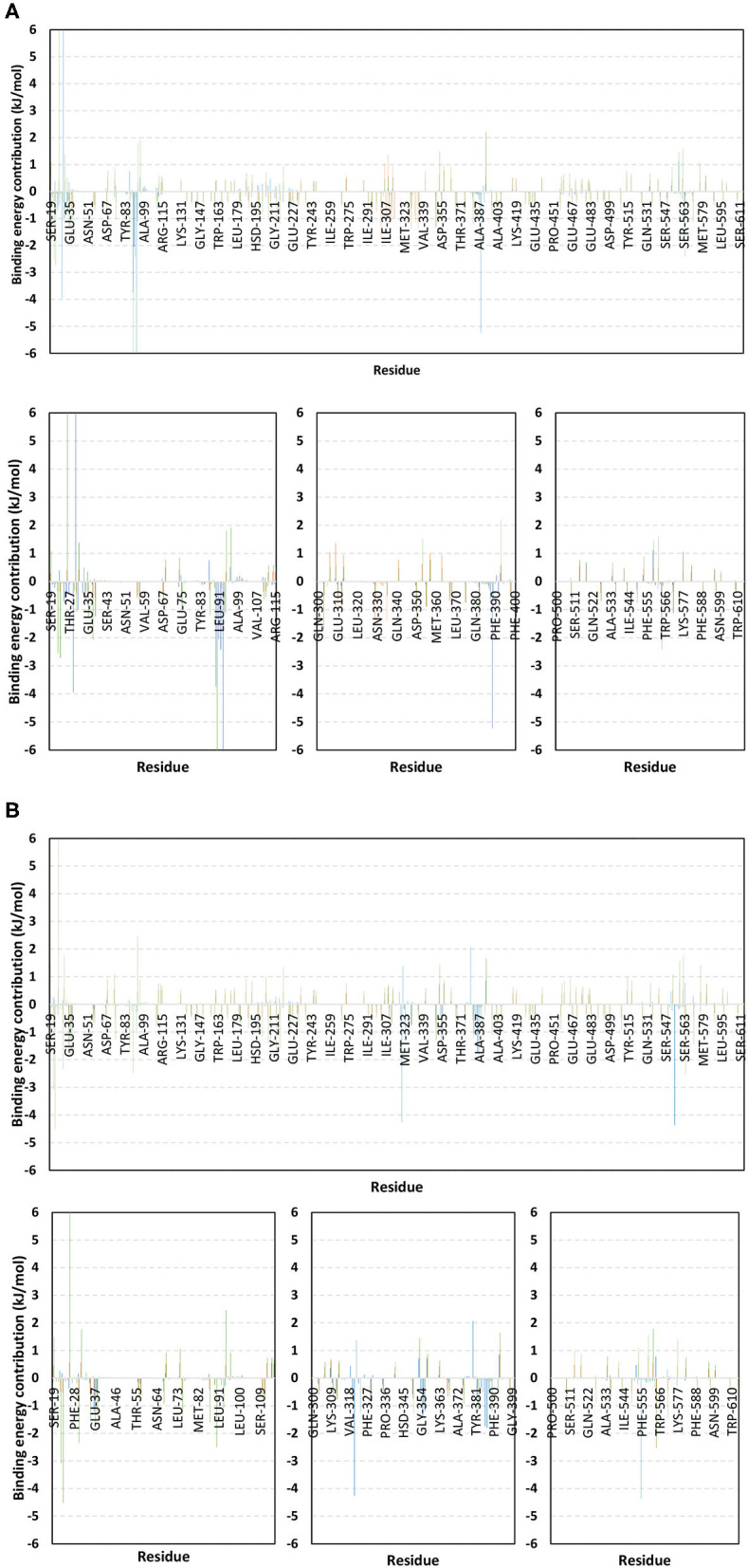
The rapid and global spread of a new human coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has produced an immediate urgency to discover promising targets for the treatment of COVID-19. Here, we consider drug repurposing as an attractive approach that can facilitate the drug discovery process by repurposing existing pharmaceuticals to treat illnesses other than their primary indications. We review current information concerning the global health issue of COVID-19 including promising approved drugs, e.g., human angiotensin-converting enzyme inhibitors (hACEIs). Besides, we describe computational approaches to be used in drug repurposing and highlight examples of in-silico studies of drug development efforts against SARS-CoV-2. Alacepril and lisinopril were found to interact with human angiotensin-converting enzyme 2 (hACE2), the host entranceway for SARS-CoV-2 spike protein, through exhibiting the most acceptable rmsd_refine values and the best binding affinity through forming a strong hydrogen bond with Asn90, which is assumed to be essential for the activity, as well as significant extra interactions with other receptor-binding residues. Furthermore, molecular dynamics (MD) simulations followed by calculation of the binding free energy were also carried out for the most promising two ligand-pocket complexes from docking studies (alacepril and lisinopril) to clarify some information on their thermodynamic and dynamic properties and confirm the docking results as well. These results we obtained probably provided an excellent lead candidate for the development of therapeutic drugs against COVID-19. Eventually, animal experiments and accurate clinical trials are needed to confirm the potential preventive and treatment effect of these compounds.
Keywords: ACEIs; COVID-19; hACE2; molecular docking; molecular dynamics.
Copyright © 2021 Al-Karmalawy, Dahab, Metwaly, Elhady, Elkaeed, Eissa and Darwish.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
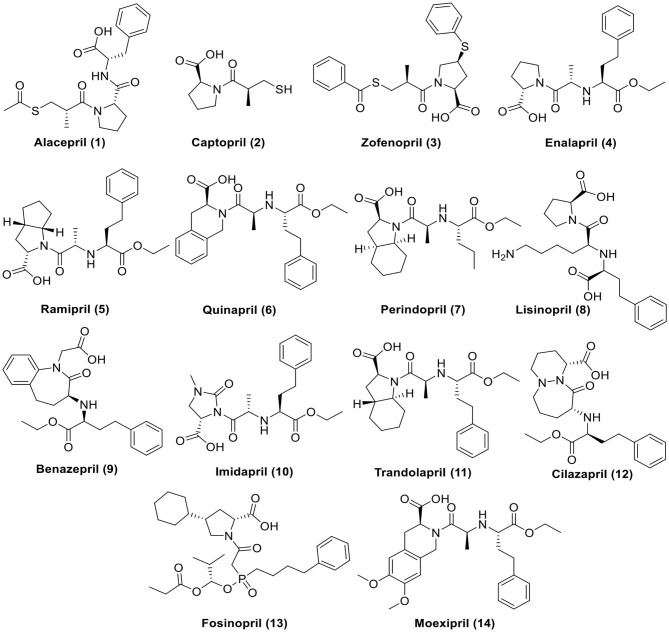

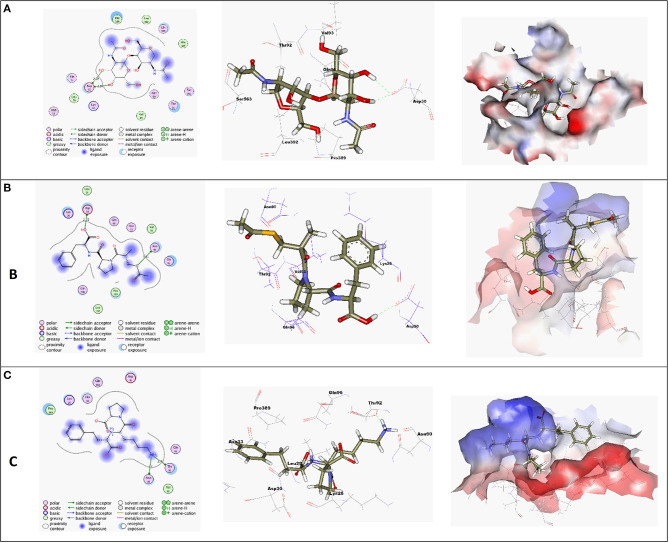
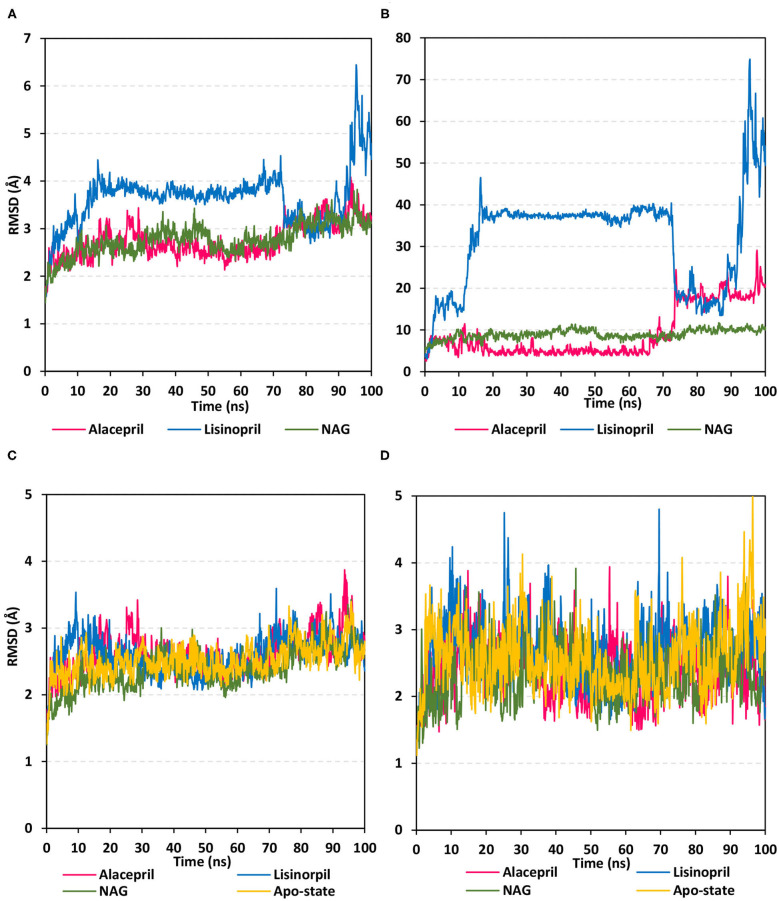
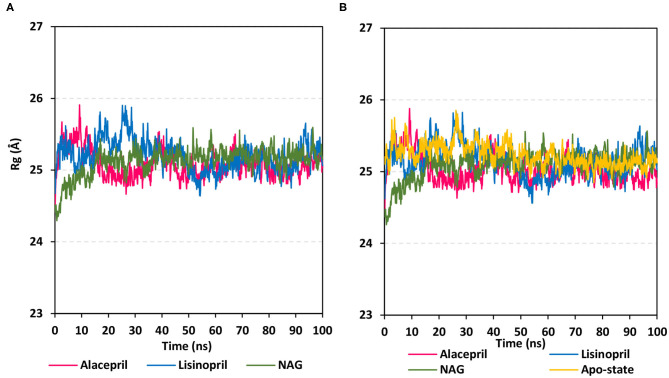
References
-
- Albuquerque S. O., Barros T. G., Dias L. R. S., Lima C. H. D. S., Azevedo P. H. R. A., Flores-Junior L. A. P., et al. . (2020). Biological evaluation and molecular modeling of peptidomimetic compounds as inhibitors for O-GlcNAc transferase (OGT). Eur. J. Pharm. Sci. 154:105510. 10.1016/j.ejps.2020.105510 - DOI - PubMed
-
- Al-Karmalawy A. A, Alnajjar R., Dahab M., Metwaly A., Eissa I. (in press). Molecular docking dynamics simulations reveal the potential of anti-HCV drugs to inhibit COVID-19 main protease. Pharm Sci. 10.34172/PS.2021.3 - DOI
-
- Al-Karmalawy A. A., Khattab M. (2020). Molecular modelling of mebendazole polymorphs as a potential colchicine binding site inhibitor. N. J. Chem. 44, 13990–13996. 10.1039/D0NJ02844D - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous