

Selection and isolation define a heterogeneous divergence landscape between hybridizing Heliconius butterflies
- PMID: 34019308
- PMCID: PMC8454027
- DOI: 10.1111/evo.14272
Selection and isolation define a heterogeneous divergence landscape between hybridizing Heliconius butterflies
Abstract
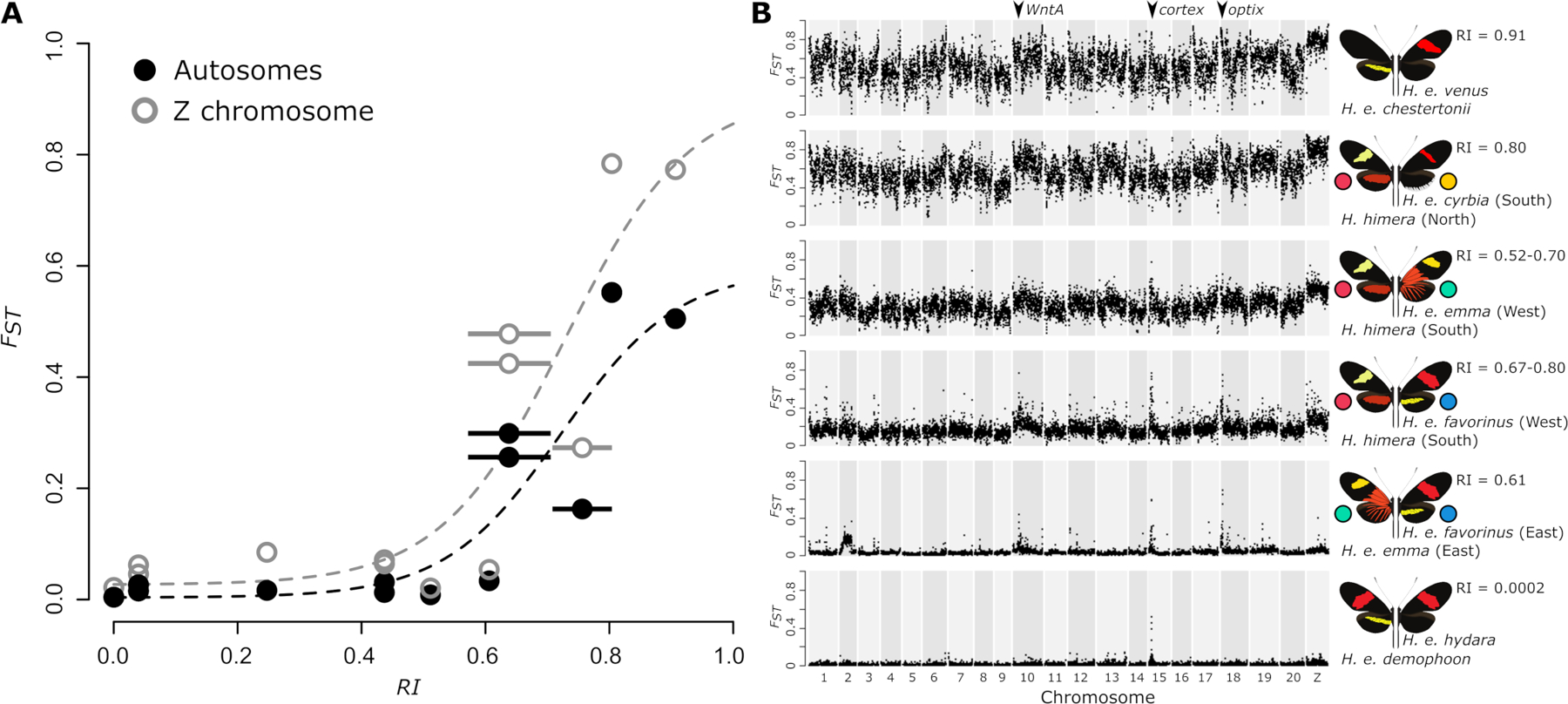
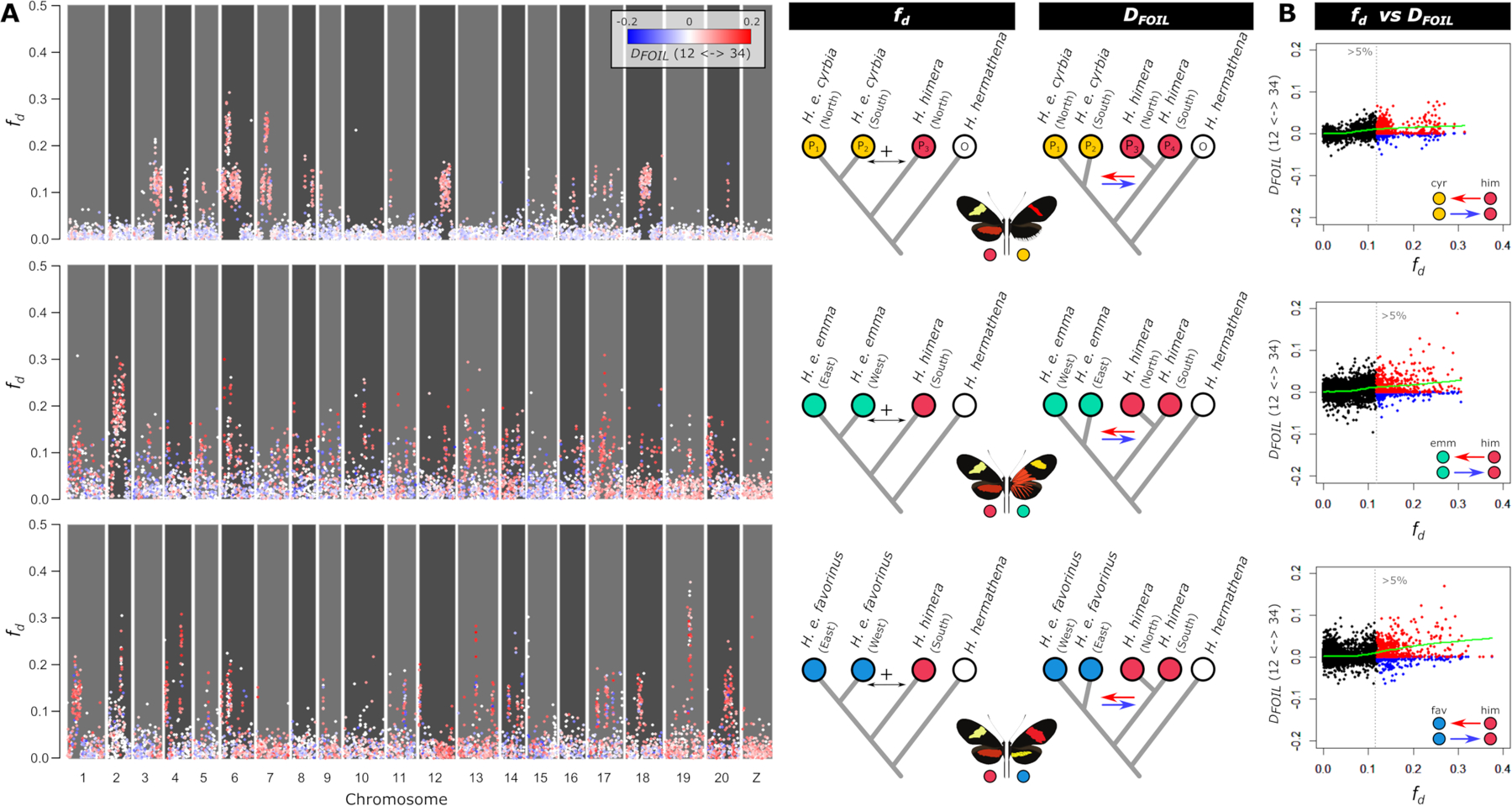
Hybridizing species provide a powerful system to identify the processes that shape genomic variation and maintain species boundaries. However, complex histories of isolation, gene flow, and selection often generate heterogeneous genomic landscapes of divergence that complicate reconstruction of the speciation history. Here, we explore patterns of divergence to reconstruct recent speciation in the erato clade of Heliconius butterflies. We focus on the genomic landscape of divergence across three contact zones of the species H. erato and H. himera. We show that these hybridizing species have an intermediate level of divergence in the erato clade, which fits with their incomplete levels of reproductive isolation. Using demographic modeling and the relationship between admixture and divergence with recombination rate variation, we reconstruct histories of gene flow, selection, and demographic change that explain the observed patterns of genomic divergence. We find that periods of isolation and selection within populations, followed by secondary contact with asymmetrical gene flow are key factors in shaping the heterogeneous genomic landscapes. Collectively, these results highlight the effectiveness of demographic modeling and recombination rate estimates to disentangling the distinct contributions of gene flow and selection to patterns of genomic divergence.
Keywords: Admixture; heterogeneous divergence; reproductive isolation; selection; species boundaries.
© 2021 The Authors. Evolution © 2021 The Society for the Study of Evolution.
Conflict of interest statement
Figures





References
-
- Arias CF, Van Belleghem S, and McMillan WO. 2016. Genomics at the evolving species boundary. Curr. Opin. Insect Sci 13:7–15. Elsevier Inc. - PubMed
-
- Barton N, and Bengtsson BO. 1986. The barrier to genetic exchange between hybridising populations. Heredity (Edinb). 57:357–376. - PubMed
-
- Barton NH, and De Cara MAR. 2009. The evolution of strong reproductive isolation. Evolution. 63:1171–1190. - PubMed
-
- Berner D, and Roesti M. 2017. Genomics of adaptive divergence with chromosome-scale heterogeneity in crossover rate. Mol. Ecol 26:6351–6369. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources