

Evaluating assembly and variant calling software for strain-resolved analysis of large DNA viruses
- PMID: 34020538
- PMCID: PMC8138829
- DOI: 10.1093/bib/bbaa123
Evaluating assembly and variant calling software for strain-resolved analysis of large DNA viruses
Abstract
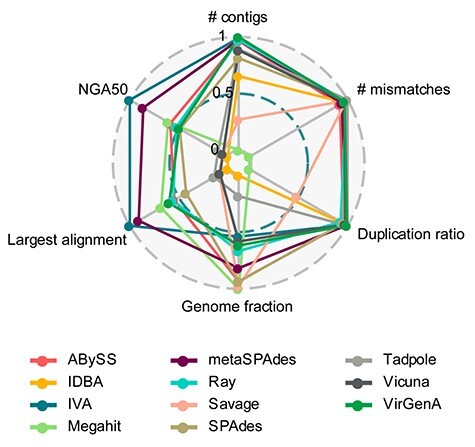
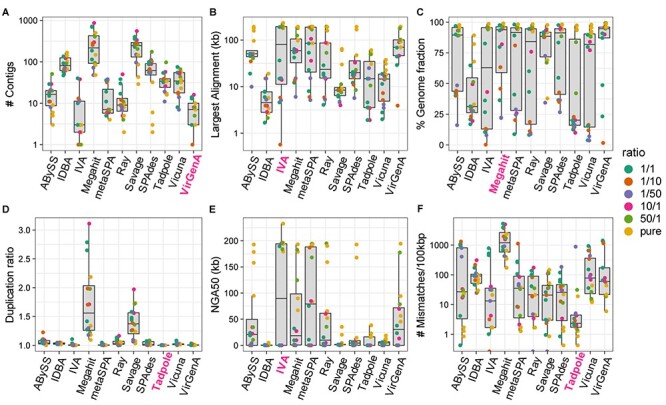
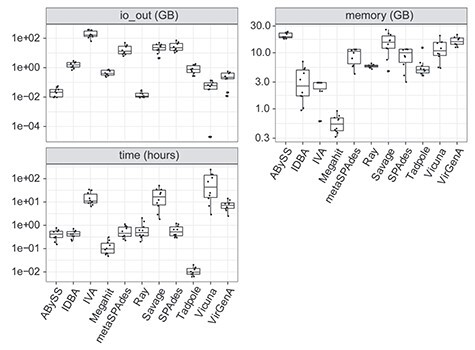
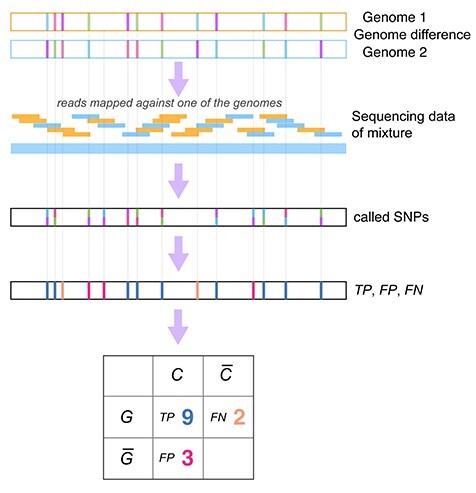
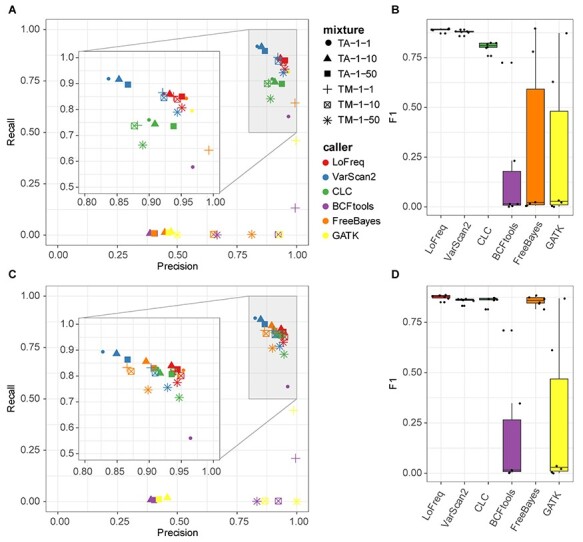
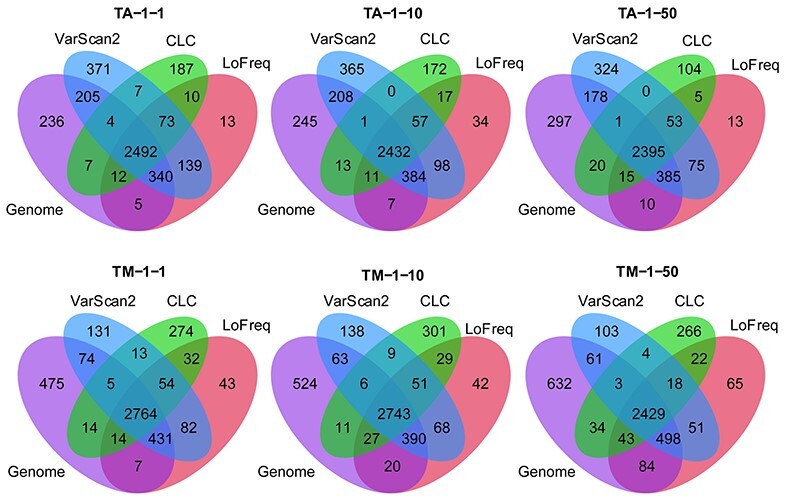
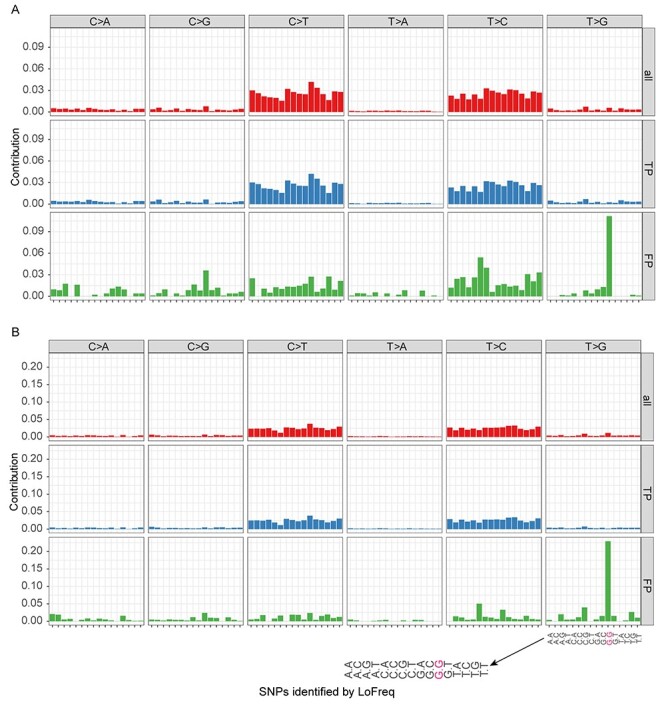
Infection with human cytomegalovirus (HCMV) can cause severe complications in immunocompromised individuals and congenitally infected children. Characterizing heterogeneous viral populations and their evolution by high-throughput sequencing of clinical specimens requires the accurate assembly of individual strains or sequence variants and suitable variant calling methods. However, the performance of most methods has not been assessed for populations composed of low divergent viral strains with large genomes, such as HCMV. In an extensive benchmarking study, we evaluated 15 assemblers and 6 variant callers on 10 lab-generated benchmark data sets created with two different library preparation protocols, to identify best practices and challenges for analyzing such data. Most assemblers, especially metaSPAdes and IVA, performed well across a range of metrics in recovering abundant strains. However, only one, Savage, recovered low abundant strains and in a highly fragmented manner. Two variant callers, LoFreq and VarScan2, excelled across all strain abundances. Both shared a large fraction of false positive variant calls, which were strongly enriched in T to G changes in a 'G.G' context. The magnitude of this context-dependent systematic error is linked to the experimental protocol. We provide all benchmarking data, results and the entire benchmarking workflow named QuasiModo, Quasispecies Metric determination on omics, under the GNU General Public License v3.0 (https://github.com/hzi-bifo/Quasimodo), to enable full reproducibility and further benchmarking on these and other data.
Keywords: HCMV; benchmark; genome assembly; strain mixtures; variant calling; virus.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Accurate assembly of full-length consensus for viral quasispecies.BMC Bioinformatics. 2025 Feb 1;26(1):36. doi: 10.1186/s12859-025-06045-z. BMC Bioinformatics. 2025. PMID: 39893441 Free PMC article.
-
Benchmarking workflows to assess performance and suitability of germline variant calling pipelines in clinical diagnostic assays.BMC Bioinformatics. 2021 Feb 24;22(1):85. doi: 10.1186/s12859-020-03934-3. BMC Bioinformatics. 2021. PMID: 33627090 Free PMC article.
-
Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection.Genome Biol. 2018 Nov 6;19(1):188. doi: 10.1186/s13059-018-1539-5. Genome Biol. 2018. PMID: 30400818 Free PMC article.
-
Best practices for evaluating single nucleotide variant calling methods for microbial genomics.Front Genet. 2015 Jul 7;6:235. doi: 10.3389/fgene.2015.00235. eCollection 2015. Front Genet. 2015. PMID: 26217378 Free PMC article. Review.
-
Genetic polymorphisms among human cytomegalovirus (HCMV) wild-type strains.Rev Med Virol. 2004 Nov-Dec;14(6):383-410. doi: 10.1002/rmv.438. Rev Med Virol. 2004. PMID: 15386592 Review.
Cited by
-
Human cytomegalovirus harnesses host L1 retrotransposon for efficient replication.Nat Commun. 2024 Sep 2;15(1):7640. doi: 10.1038/s41467-024-51961-y. Nat Commun. 2024. PMID: 39223139 Free PMC article.
-
High-throughput engineering of cytoplasmic- and nuclear-replicating large dsDNA viruses by CRISPR/Cas9.J Gen Virol. 2022 Oct;103(10):001797. doi: 10.1099/jgv.0.001797. J Gen Virol. 2022. PMID: 36260063 Free PMC article.
-
Grapevine Virology in the Third-Generation Sequencing Era: From Virus Detection to Viral Epitranscriptomics.Plants (Basel). 2021 Oct 31;10(11):2355. doi: 10.3390/plants10112355. Plants (Basel). 2021. PMID: 34834718 Free PMC article. Review.
-
Strain-resolved de-novo metagenomic assembly of viral genomes and microbial 16S rRNAs.Microbiome. 2024 Oct 1;12(1):187. doi: 10.1186/s40168-024-01904-y. Microbiome. 2024. PMID: 39354646 Free PMC article.
-
Identifying high-confidence variants in human cytomegalovirus genomes sequenced from clinical samples.Virus Evol. 2022 Dec 5;8(2):veac114. doi: 10.1093/ve/veac114. eCollection 2022. Virus Evol. 2022. PMID: 37091479 Free PMC article.
References
-
- Griffiths P, Baraniak I, Reeves M. The pathogenesis of human cytomegalovirus. J Pathol 2015;235:288–97. - PubMed
-
- Dolan A, Cunningham C, Hector RD, et al. Genetic content of wild-type human cytomegalovirus. J Gen Virol 2004;85:1301–12. - PubMed
-
- Campillo-Balderas JA, Lazcano A, Becerra A. Viral genome size distribution does not correlate with the antiquity of the host lineages. Front Ecol Evol 2015;3:728.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
