

A comprehensive survey of regulatory network inference methods using single cell RNA sequencing data
- PMID: 34020546
- PMCID: PMC8138892
- DOI: 10.1093/bib/bbaa190
A comprehensive survey of regulatory network inference methods using single cell RNA sequencing data
Abstract
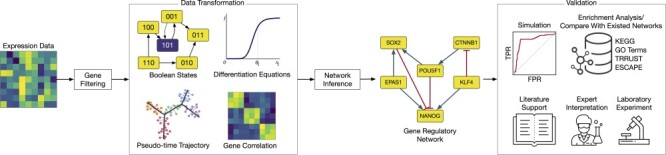
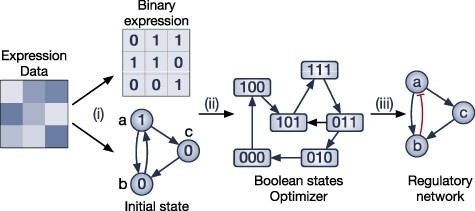
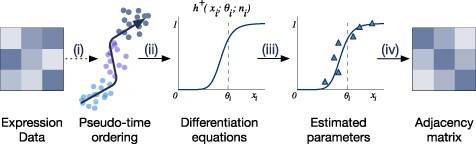
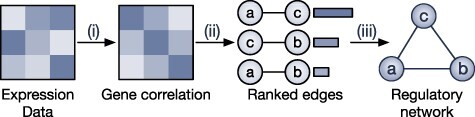
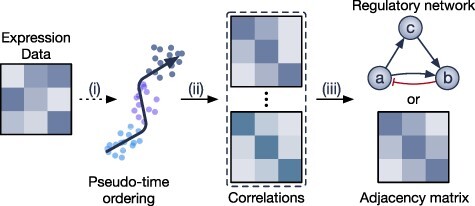
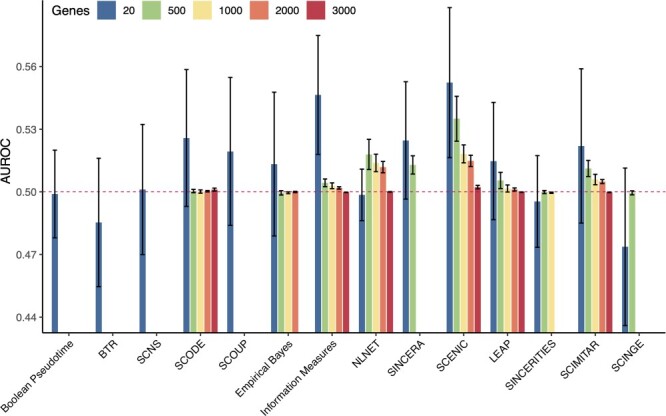
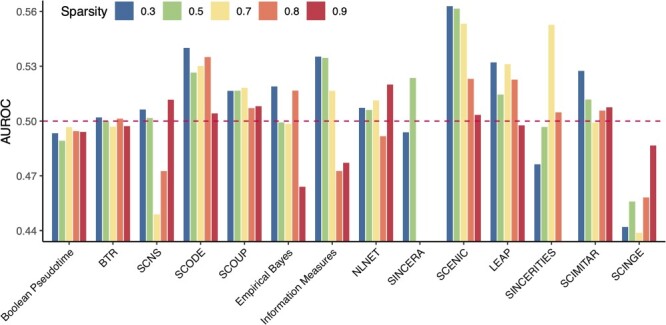
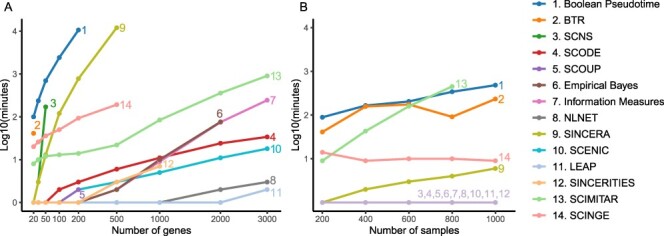
Gene regulatory network is a complicated set of interactions between genetic materials, which dictates how cells develop in living organisms and react to their surrounding environment. Robust comprehension of these interactions would help explain how cells function as well as predict their reactions to external factors. This knowledge can benefit both developmental biology and clinical research such as drug development or epidemiology research. Recently, the rapid advance of single-cell sequencing technologies, which pushed the limit of transcriptomic profiling to the individual cell level, opens up an entirely new area for regulatory network research. To exploit this new abundant source of data and take advantage of data in single-cell resolution, a number of computational methods have been proposed to uncover the interactions hidden by the averaging process in standard bulk sequencing. In this article, we review 15 such network inference methods developed for single-cell data. We discuss their underlying assumptions, inference techniques, usability, and pros and cons. In an extensive analysis using simulation, we also assess the methods' performance, sensitivity to dropout and time complexity. The main objective of this survey is to assist not only life scientists in selecting suitable methods for their data and analysis purposes but also computational scientists in developing new methods by highlighting outstanding challenges in the field that remain to be addressed in the future development.
Keywords: RNA sequencing; gene regulatory network; scRNA-seq; simulation studies; single-cell data.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Microbiol 2010; 8(10): 717–29. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
