

Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy
- PMID: 34020912
- PMCID: PMC9064286
- DOI: 10.1016/j.trecan.2021.04.003
Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy
Abstract
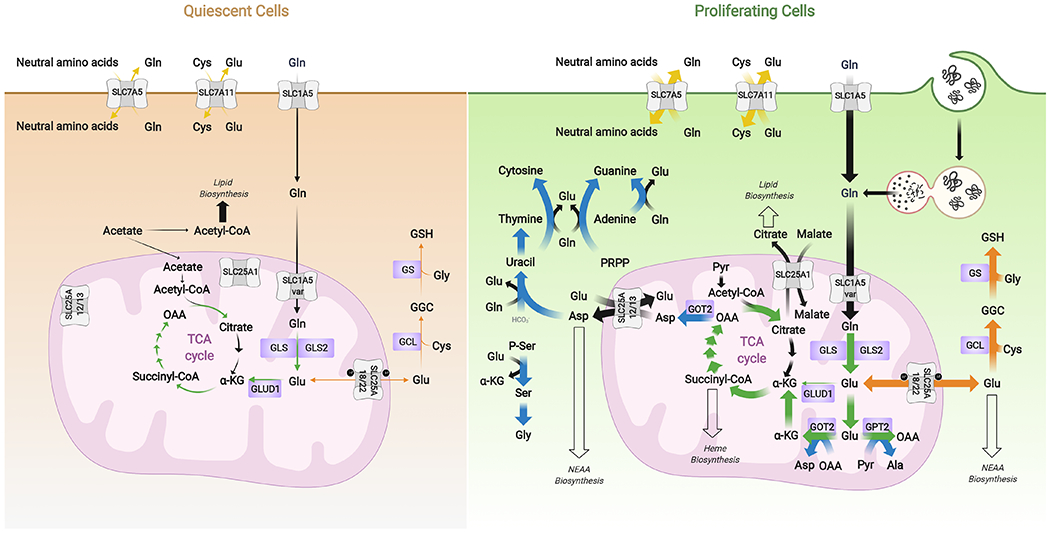
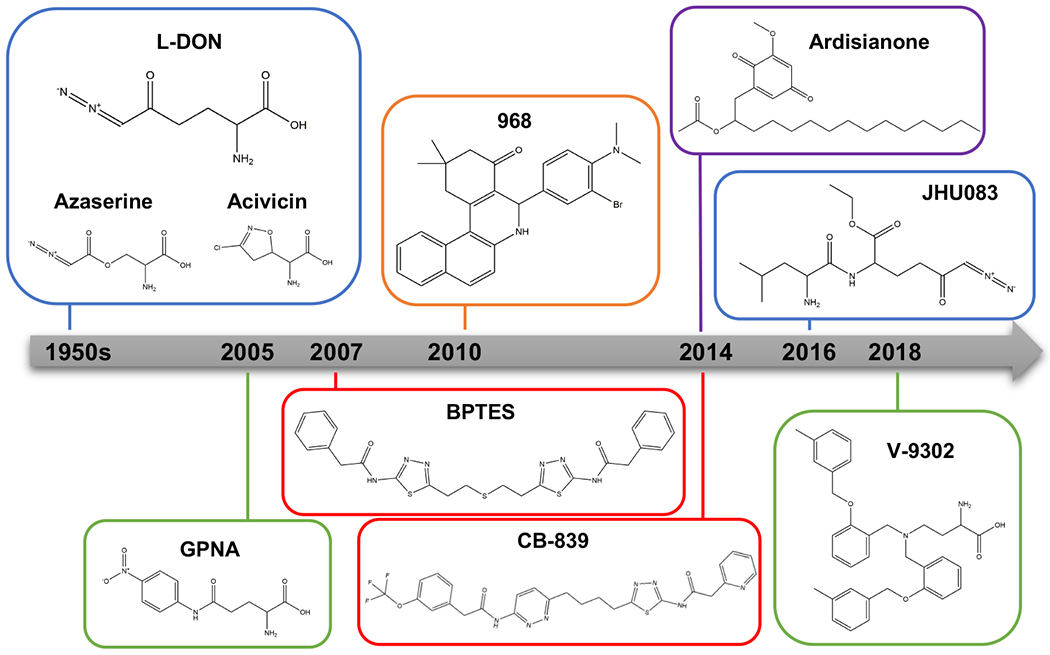
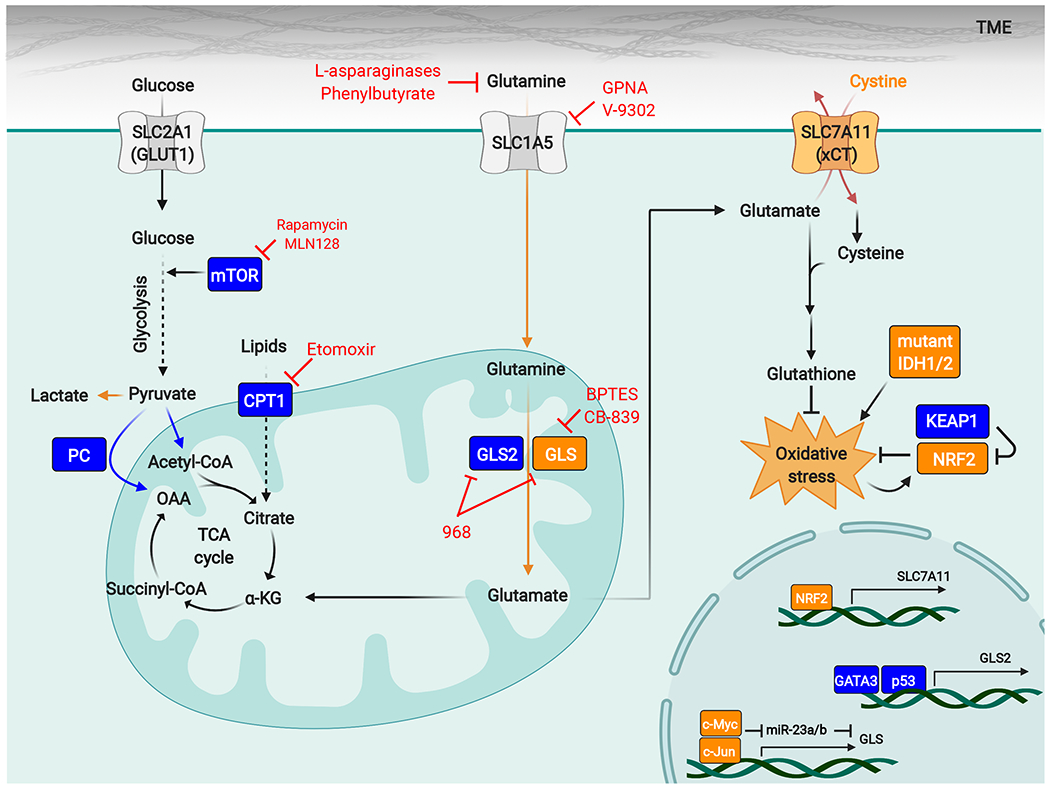
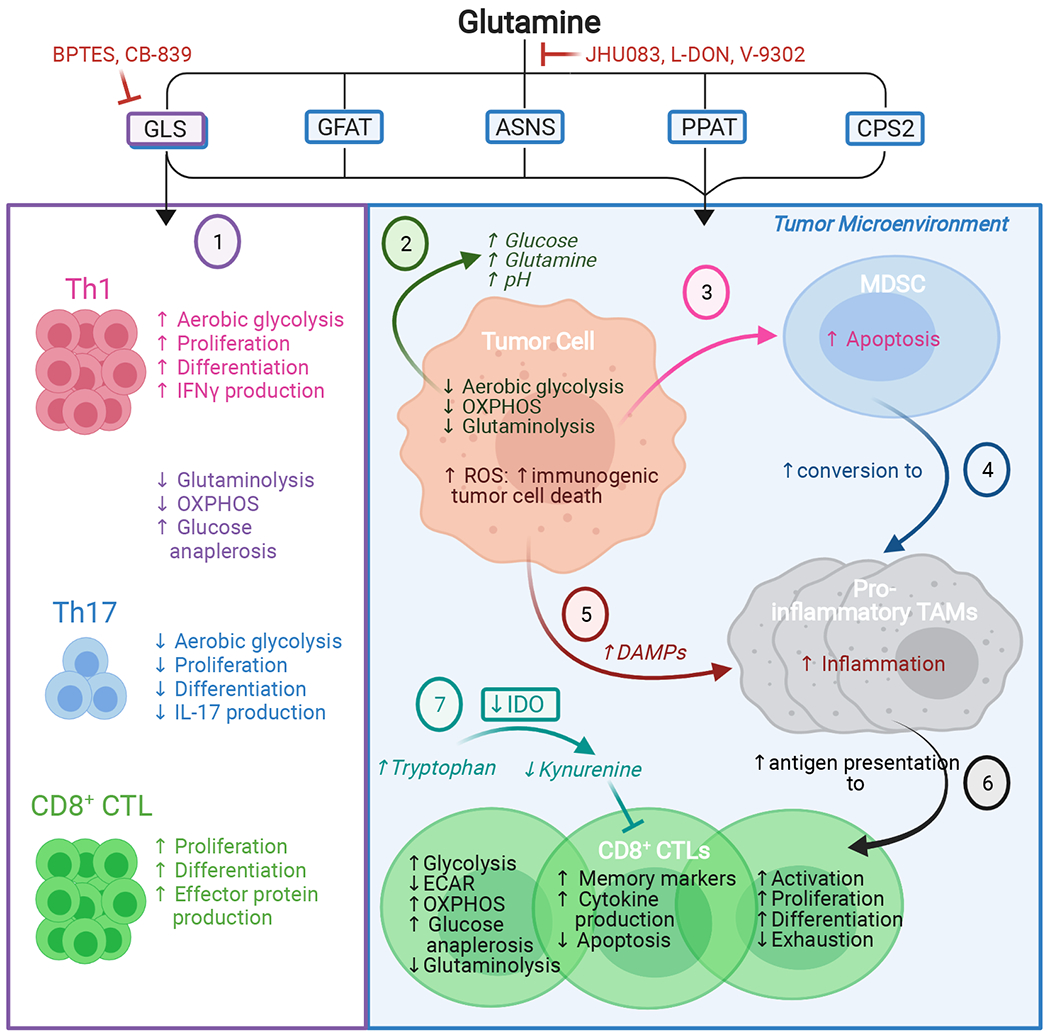
Glutamine metabolism is reprogrammed during tumorigenesis and has been investigated as a promising target for cancer therapy. However, efforts to drug this process are confounded by the intrinsic metabolic heterogeneity and flexibility of tumors, as well as the risk of adverse effects on the anticancer immune response. Recent research has yielded important insights into the mechanisms that determine the tumor and the host immune responses to pharmacological perturbation of glutamine metabolism. Here, we discuss these findings and suggest that, collectively, they point toward patient stratification and drug combination strategies to maximize the efficacy of glutamine metabolism inhibitors as cancer therapeutics.
Keywords: CB-839; JHU083; cancer metabolism; glutaminase; glutamine; immunometabolism.
Copyright © 2021 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism.J Biol Chem. 2018 Mar 9;293(10):3535-3545. doi: 10.1074/jbc.M117.810101. Epub 2018 Jan 9. J Biol Chem. 2018. PMID: 29317493 Free PMC article.
-
Glutamine Metabolism Heterogeneity in Glioblastoma Unveils an Innovative Combination Therapy Strategy.J Mol Neurosci. 2024 May 10;74(2):52. doi: 10.1007/s12031-024-02201-x. J Mol Neurosci. 2024. PMID: 38724832
-
Pyruvate anaplerosis is a mechanism of resistance to pharmacological glutaminase inhibition in triple-receptor negative breast cancer.BMC Cancer. 2020 May 25;20(1):470. doi: 10.1186/s12885-020-06885-3. BMC Cancer. 2020. PMID: 32450839 Free PMC article.
-
Targeting GLS1 to cancer therapy through glutamine metabolism.Clin Transl Oncol. 2021 Nov;23(11):2253-2268. doi: 10.1007/s12094-021-02645-2. Epub 2021 May 23. Clin Transl Oncol. 2021. PMID: 34023970 Review.
-
Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer.Semin Cell Dev Biol. 2020 Feb;98:34-43. doi: 10.1016/j.semcdb.2019.05.012. Epub 2019 May 22. Semin Cell Dev Biol. 2020. PMID: 31100352 Review.
Cited by
-
Molecular mechanisms of cisplatin resistance in ovarian cancer.Genes Dis. 2023 Aug 2;11(6):101063. doi: 10.1016/j.gendis.2023.06.032. eCollection 2024 Nov. Genes Dis. 2023. PMID: 39224110 Free PMC article. Review.
-
Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity.J Biol Chem. 2022 Apr;298(4):101753. doi: 10.1016/j.jbc.2022.101753. Epub 2022 Feb 19. J Biol Chem. 2022. PMID: 35189139 Free PMC article.
-
Metabolic partitioning in the brain and its hijacking by glioblastoma.Genes Dev. 2023 Aug 1;37(15-16):681-702. doi: 10.1101/gad.350693.123. Epub 2023 Aug 30. Genes Dev. 2023. PMID: 37648371 Free PMC article. Review.
-
Amino acid metabolism reprogramming: shedding new light on T cell anti-tumor immunity.J Exp Clin Cancer Res. 2023 Nov 3;42(1):291. doi: 10.1186/s13046-023-02845-4. J Exp Clin Cancer Res. 2023. PMID: 37924140 Free PMC article. Review.
-
High glutamine increases stroke risk by inducing the endothelial-to-mesenchymal transition in moyamoya disease.MedComm (2020). 2024 Apr 15;5(5):e525. doi: 10.1002/mco2.525. eCollection 2024 May. MedComm (2020). 2024. PMID: 38628905 Free PMC article.
References
-
- Warburg O (1925) The metabolism of carcinoma cells. The Journal of Cancer Research 9, 148–163.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
