

Overarching control of autophagy and DNA damage response by CHD6 revealed by modeling a rare human pathology
- PMID: 34021162
- PMCID: PMC8140133
- DOI: 10.1038/s41467-021-23327-1
Overarching control of autophagy and DNA damage response by CHD6 revealed by modeling a rare human pathology
Abstract
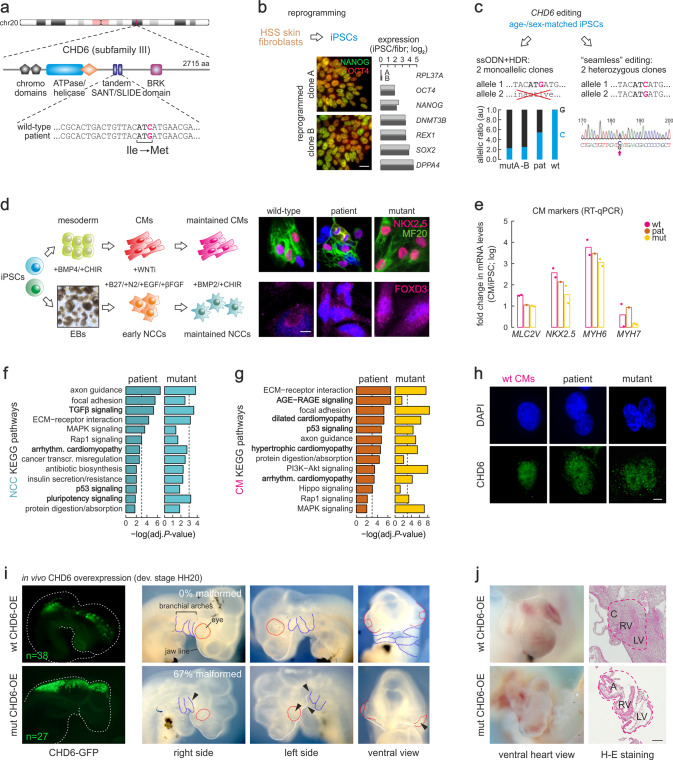
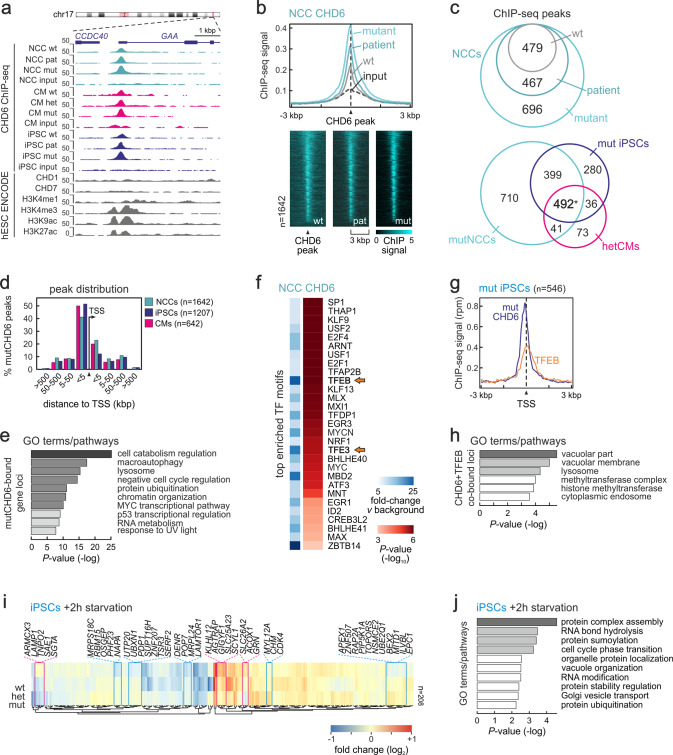
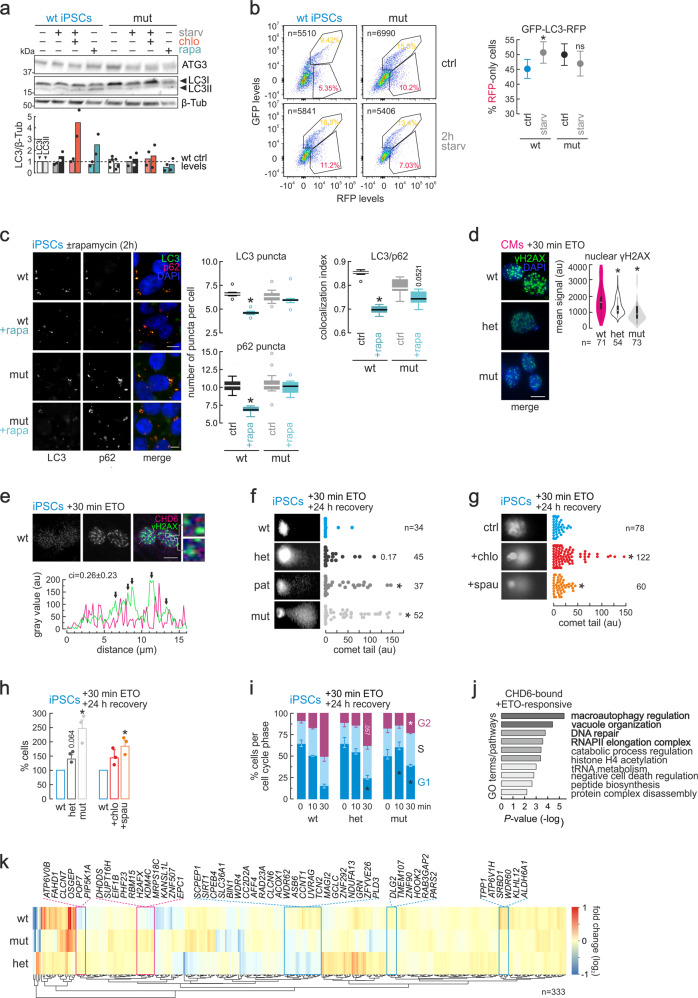
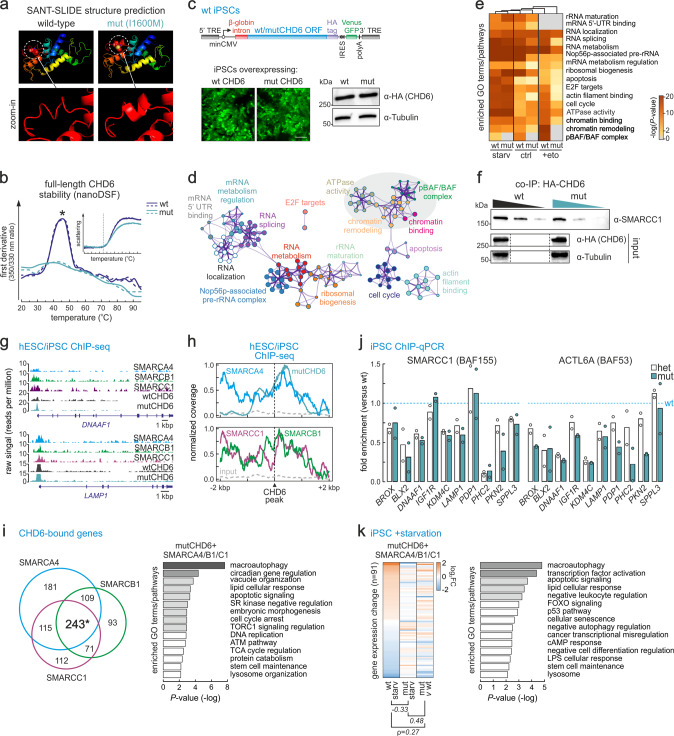
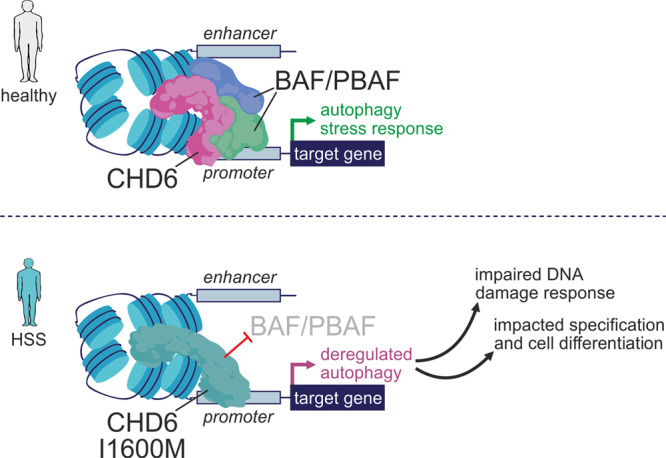
Members of the chromodomain-helicase-DNA binding (CHD) protein family are chromatin remodelers implicated in human pathologies, with CHD6 being one of its least studied members. We discovered a de novo CHD6 missense mutation in a patient clinically presenting the rare Hallermann-Streiff syndrome (HSS). We used genome editing to generate isogenic iPSC lines and model HSS in relevant cell types. By combining genomics with functional in vivo and in vitro assays, we show that CHD6 binds a cohort of autophagy and stress response genes across cell types. The HSS mutation affects CHD6 protein folding and impairs its ability to recruit co-remodelers in response to DNA damage or autophagy stimulation. This leads to accumulation of DNA damage burden and senescence-like phenotypes. We therefore uncovered a molecular mechanism explaining HSS onset via chromatin control of autophagic flux and genotoxic stress surveillance.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
CHD6 promotes broad nucleosome eviction for transcriptional activation in prostate cancer cells.Nucleic Acids Res. 2022 Nov 28;50(21):12186-12201. doi: 10.1093/nar/gkac1090. Nucleic Acids Res. 2022. PMID: 36408932 Free PMC article.
-
The CHD6 chromatin remodeler is an oxidative DNA damage response factor.Nat Commun. 2019 Jan 16;10(1):241. doi: 10.1038/s41467-018-08111-y. Nat Commun. 2019. PMID: 30651562 Free PMC article.
-
The ATP-dependent chromatin remodeling enzymes CHD6, CHD7, and CHD8 exhibit distinct nucleosome binding and remodeling activities.J Biol Chem. 2017 Jul 14;292(28):11927-11936. doi: 10.1074/jbc.M117.779470. Epub 2017 May 21. J Biol Chem. 2017. PMID: 28533432 Free PMC article.
-
The Chromodomain Helicase DNA-Binding Chromatin Remodelers: Family Traits that Protect from and Promote Cancer.Cold Spring Harb Perspect Med. 2017 Apr 3;7(4):a026450. doi: 10.1101/cshperspect.a026450. Cold Spring Harb Perspect Med. 2017. PMID: 28096241 Free PMC article. Review.
-
CHD chromatin remodelling enzymes and the DNA damage response.Mutat Res. 2013 Oct;750(1-2):31-44. doi: 10.1016/j.mrfmmm.2013.07.008. Epub 2013 Aug 14. Mutat Res. 2013. PMID: 23954449 Review.
Cited by
-
Remodeling of the endothelial cell transcriptional program via paracrine and DNA-binding activities of MPO.iScience. 2024 Jan 12;27(2):108898. doi: 10.1016/j.isci.2024.108898. eCollection 2024 Feb 16. iScience. 2024. PMID: 38322992 Free PMC article.
-
Hydroxygenkwanin Increases the Sensitivity of Liver Cancer Cells to Chemotherapy by Inhibiting DNA Damage Response in Mouse Xenograft Models.Int J Mol Sci. 2021 Sep 9;22(18):9766. doi: 10.3390/ijms22189766. Int J Mol Sci. 2021. PMID: 34575923 Free PMC article.
-
CHD6 promotes broad nucleosome eviction for transcriptional activation in prostate cancer cells.Nucleic Acids Res. 2022 Nov 28;50(21):12186-12201. doi: 10.1093/nar/gkac1090. Nucleic Acids Res. 2022. PMID: 36408932 Free PMC article.
-
The chromatin remodeler CHD6 promotes colorectal cancer development by regulating TMEM65-mediated mitochondrial dynamics via EGF and Wnt signaling.Cell Discov. 2022 Dec 6;8(1):130. doi: 10.1038/s41421-022-00478-z. Cell Discov. 2022. PMID: 36473865 Free PMC article.
-
Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins.Genes (Basel). 2021 Nov 19;12(11):1827. doi: 10.3390/genes12111827. Genes (Basel). 2021. PMID: 34828433 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
