

Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities
- PMID: 34021239
- PMCID: PMC8139981
- DOI: 10.1038/s42003-021-02112-2
Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities
Abstract
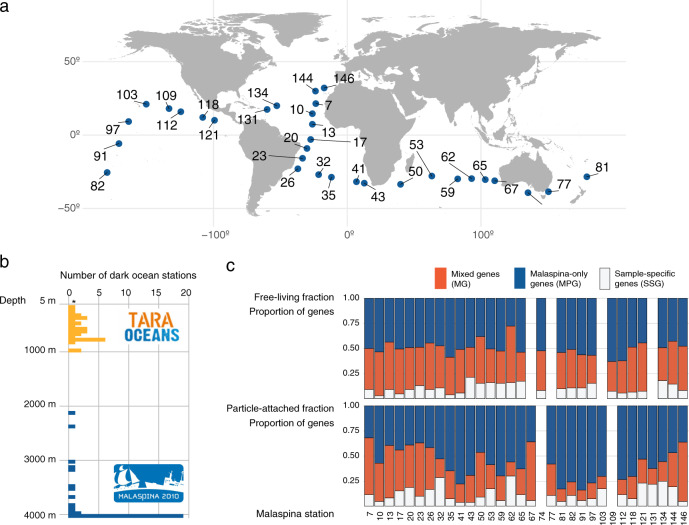
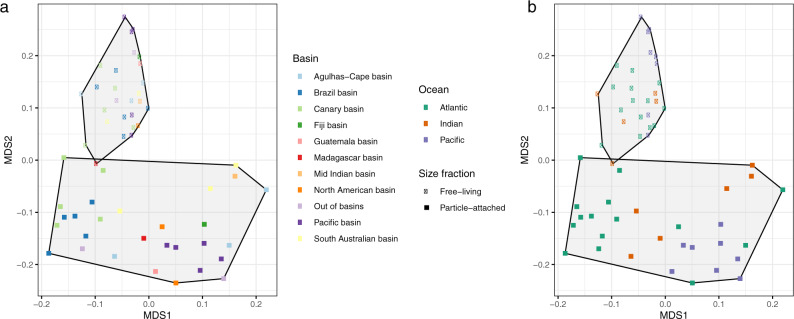
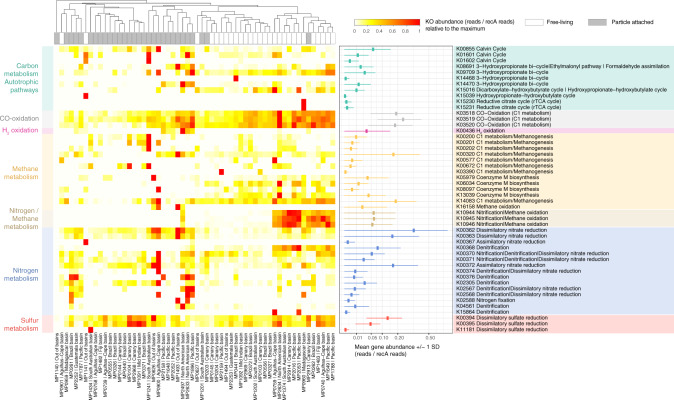
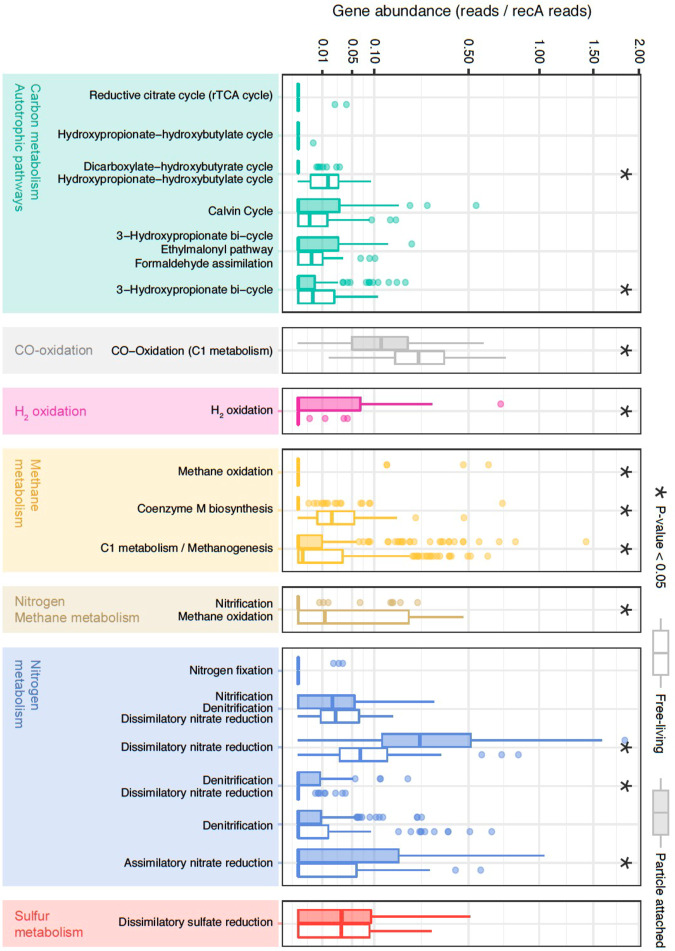
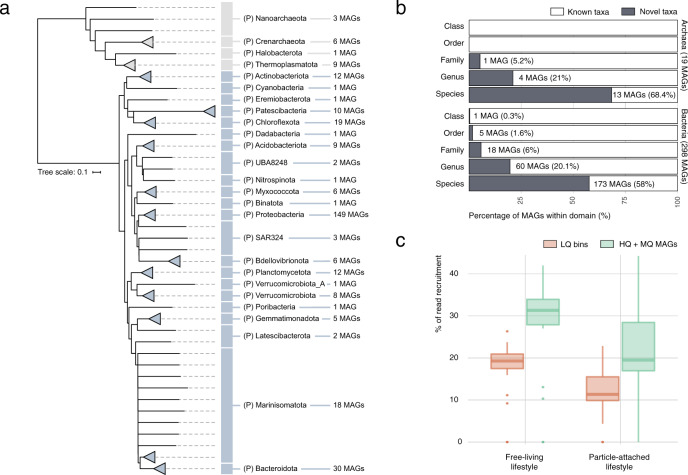
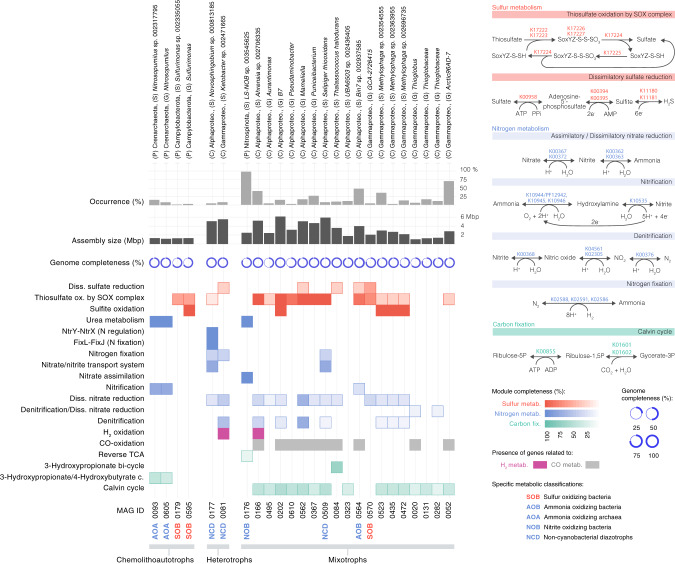
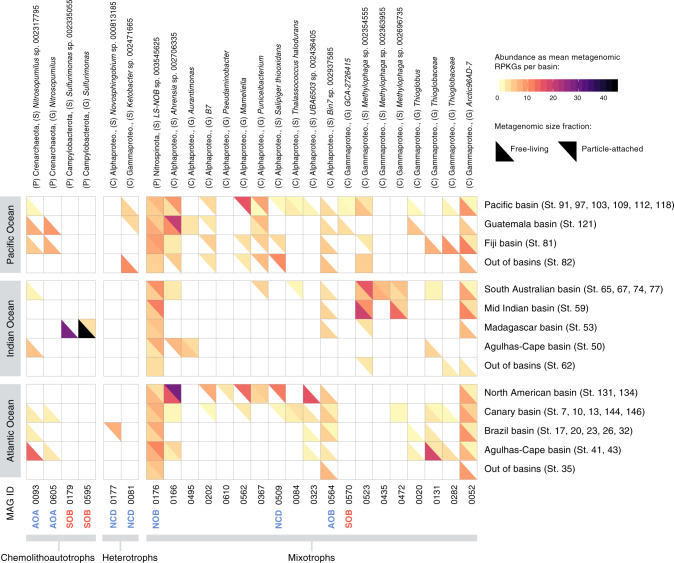
The deep sea, the largest ocean's compartment, drives planetary-scale biogeochemical cycling. Yet, the functional exploration of its microbial communities lags far behind other environments. Here we analyze 58 metagenomes from tropical and subtropical deep oceans to generate the Malaspina Gene Database. Free-living or particle-attached lifestyles drive functional differences in bathypelagic prokaryotic communities, regardless of their biogeography. Ammonia and CO oxidation pathways are enriched in the free-living microbial communities and dissimilatory nitrate reduction to ammonium and H2 oxidation pathways in the particle-attached, while the Calvin Benson-Bassham cycle is the most prevalent inorganic carbon fixation pathway in both size fractions. Reconstruction of the Malaspina Deep Metagenome-Assembled Genomes reveals unique non-cyanobacterial diazotrophic bacteria and chemolithoautotrophic prokaryotes. The widespread potential to grow both autotrophically and heterotrophically suggests that mixotrophy is an ecologically relevant trait in the deep ocean. These results expand our understanding of the functional microbial structure and metabolic capabilities of the largest Earth aquatic ecosystem.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Cho BC, Azam F. major role of bacteria in biogeochemical fluxes in the ocean´s interior. Nature. 1988;332:441–443. doi: 10.1038/332441a0. - DOI
-
- Aristegui J, Gasol JM, Duarte CM, Herndl GJ. Microbial oceanography of the dark ocean’s pelagic realm. Limnol. Oceanogr. 2009;54:1501–1529. doi: 10.4319/lo.2009.54.5.1501. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
