

Non-coding region variants upstream of MEF2C cause severe developmental disorder through three distinct loss-of-function mechanisms
- PMID: 34022131
- PMCID: PMC8206381
- DOI: 10.1016/j.ajhg.2021.04.025
Non-coding region variants upstream of MEF2C cause severe developmental disorder through three distinct loss-of-function mechanisms
Abstract
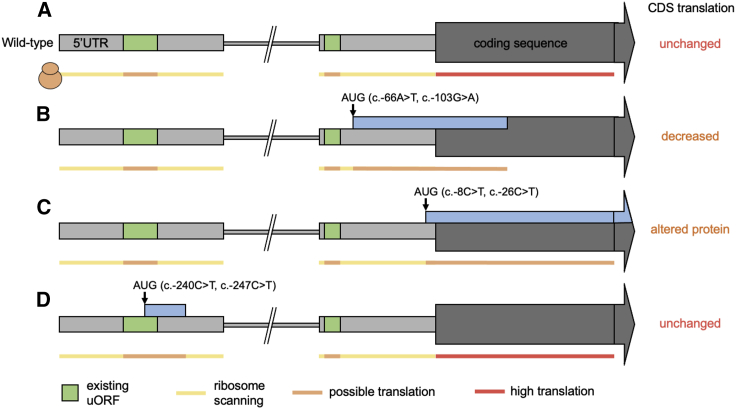
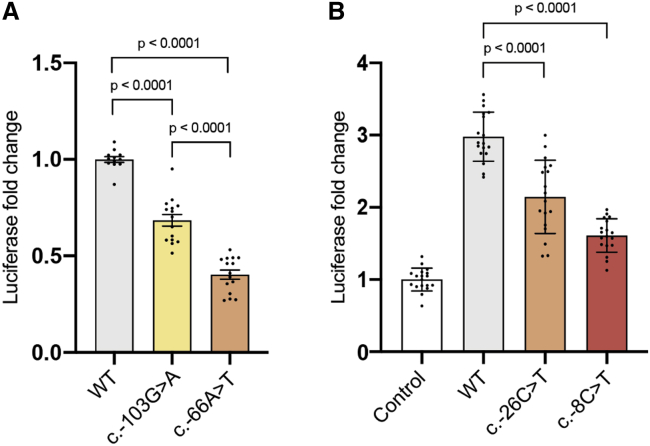
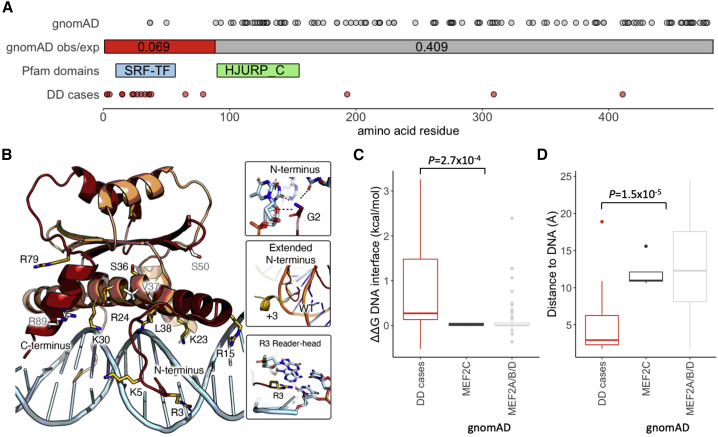
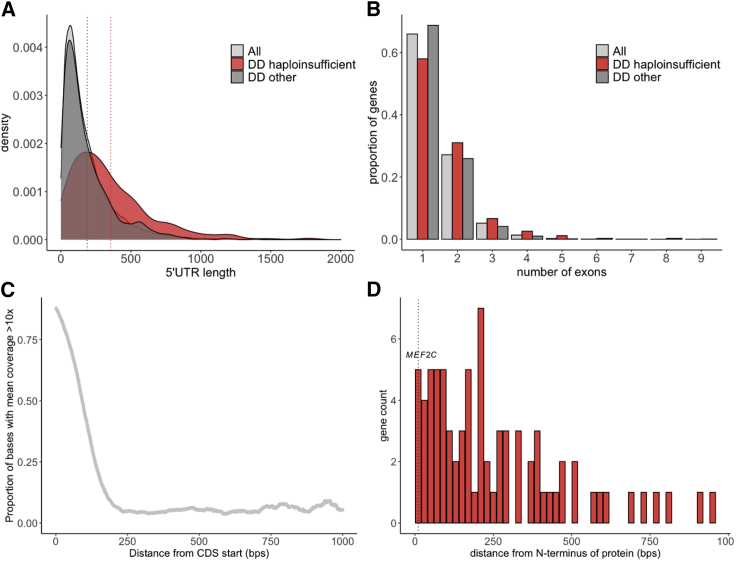
Clinical genetic testing of protein-coding regions identifies a likely causative variant in only around half of developmental disorder (DD) cases. The contribution of regulatory variation in non-coding regions to rare disease, including DD, remains very poorly understood. We screened 9,858 probands from the Deciphering Developmental Disorders (DDD) study for de novo mutations in the 5' untranslated regions (5' UTRs) of genes within which variants have previously been shown to cause DD through a dominant haploinsufficient mechanism. We identified four single-nucleotide variants and two copy-number variants upstream of MEF2C in a total of ten individual probands. We developed multiple bespoke and orthogonal experimental approaches to demonstrate that these variants cause DD through three distinct loss-of-function mechanisms, disrupting transcription, translation, and/or protein function. These non-coding region variants represent 23% of likely diagnoses identified in MEF2C in the DDD cohort, but these would all be missed in standard clinical genetics approaches. Nonetheless, these variants are readily detectable in exome sequence data, with 30.7% of 5' UTR bases across all genes well covered in the DDD dataset. Our analyses show that non-coding variants upstream of genes within which coding variants are known to cause DD are an important cause of severe disease and demonstrate that analyzing 5' UTRs can increase diagnostic yield. We also show how non-coding variants can help inform both the disease-causing mechanism underlying protein-coding variants and dosage tolerance of the gene.
Keywords: developmental disorders, clinical genetic testing, non-coding region variants, 5' UTR variants.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
K.J.K. is a consultant for Vor Biopharma. J.J. and K.R. are employees of GeneDx, Inc. K.R. holds shares in Opko Health, Inc. B.D.Z. is a member of the speakers bureau for Biogen, Neurelis, and Supernus. S.A.C. is co-founder and shareholder of Enleofen Bio Pte Ltd. M.E.H. is co-founder, shareholder, consultant, and non-executive director of Congenica Ltd. All other authors declare no competing interests.
Figures
References
-
- Spielmann M., Mundlos S. Looking beyond the genes: the role of non-coding variants in human disease. Hum. Mol. Genet. 2016;25(R2):R157–R165. - PubMed
-
- Chong J.X., Buckingham K.J., Jhangiani S.N., Boehm C., Sobreira N., Smith J.D., Harrell T.M., McMillin M.J., Wiszniewski W., Gambin T., Centers for Mendelian Genomics The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015;97:199–215. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
