

The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: a systematic literature review
- PMID: 34022943
- PMCID: PMC8141220
- DOI: 10.1186/s13023-021-01862-w
The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: a systematic literature review
Abstract
Background: Duchenne muscular dystrophy (DMD) is a severe rare progressive inherited neuromuscular disorder, leading to loss of ambulation (LOA) and premature mortality. The standard of care for patients with DMD has been treatment with corticosteroids for the past decade; however a synthesis of contemporary data describing the clinical course of DMD is lacking. The objective was to summarize age at key clinical milestones (loss of ambulation, scoliosis, ventilation, cardiomyopathy, and mortality) in the corticosteroid-treatment-era.
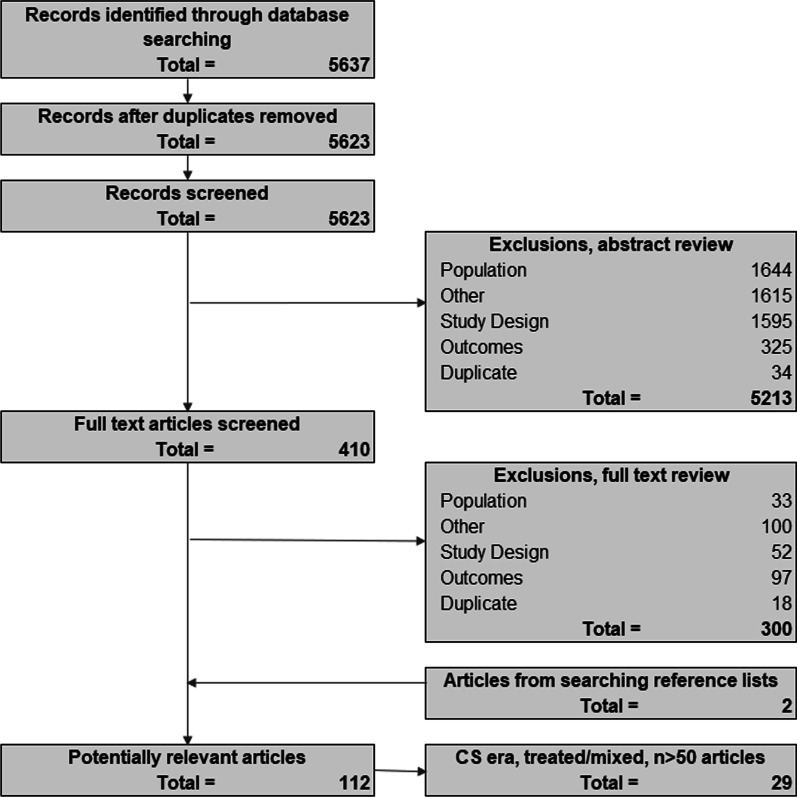
Methods: A systematic review was conducted using MEDLINE and EMBASE. The percentage experiencing key clinical milestones, and the mean or median age at those milestones, was synthesized from studies from North American populations, published between 2007 and 2018.
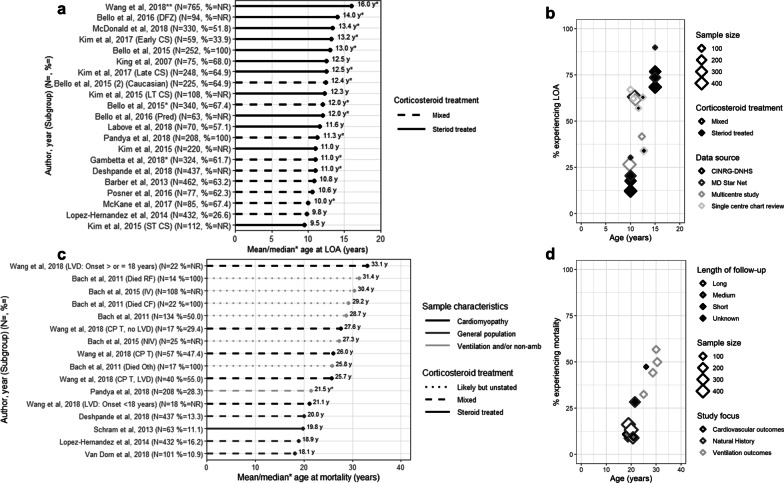
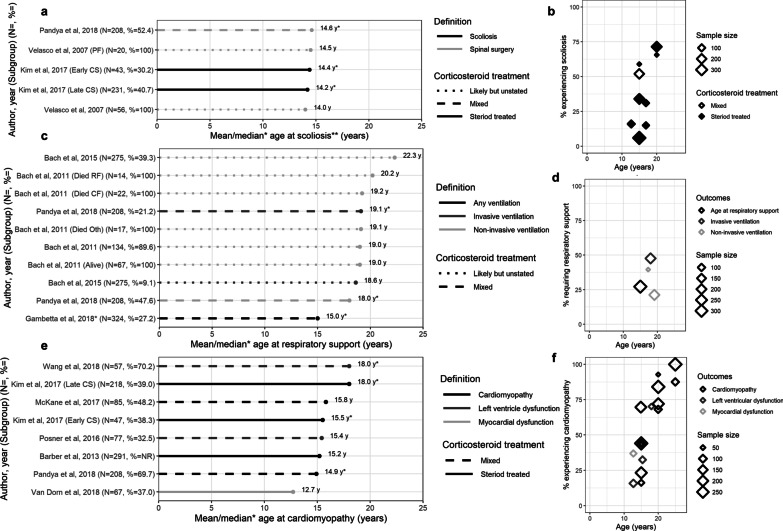
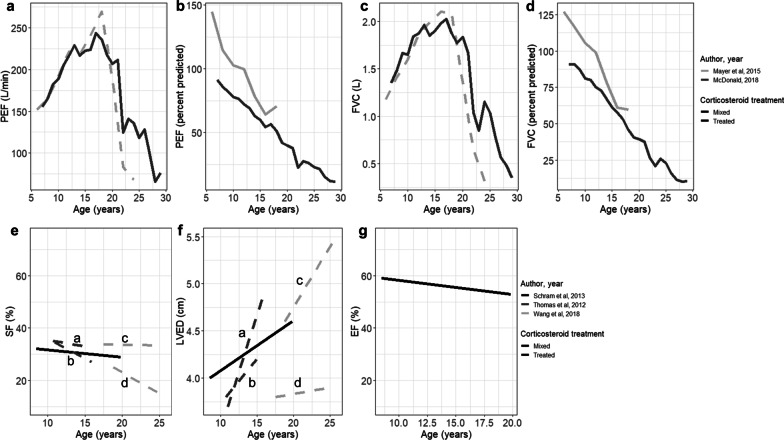
Results: From 5637 abstracts, 29 studies were included. Estimates of the percentage experiencing key clinical milestones, and age at those milestones, showed heterogeneity. Up to 30% of patients lost ambulation by age 10 years, and up to 90% by 15 years of age. The mean age at scoliosis onset was approximately 14 years. Ventilatory support began from 15 to 18 years, and up to half of patients required ventilation by 20 years of age. Registry-based estimates suggest that 70% had evidence of cardiomyopathy by 15 years and almost all by 20 years of age. Finally, mortality rates up to 16% by age 20 years were reported; among those surviving to adulthood mortality was up to 60% by age 30 years.
Conclusions: Contemporary natural history studies from North America report that LOA on average occurs in the early teens, need for ventilation and cardiomyopathy in the late teens, and death in the third or fourth decade of life. Variability in rates may be due to differences in study design, treatment with corticosteroids or other disease-modifying agents, variations in clinical practices, and dystrophin mutations. Despite challenges in synthesizing estimates, these findings help characterize disease progression among contemporary North American DMD patients.
Keywords: Clinical course; DMD; Duchenne muscular dystrophy; Loss of ambulation; Systematic review.
Conflict of interest statement
SMS and AMD are employees of Broadstreet HEOR, and MH was at the time of this project; which received funds from Sarepta for this work. RMS and KLG are employees of Sarepta. JM acted as a consultant to Broadstreet HEOR.
Figures
References
-
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–267. doi: 10.1016/S1474-4422(18)30024-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
