

Meanders as a scaling motif for understanding of floodplain soil microbiome and biogeochemical potential at the watershed scale
- PMID: 34022966
- PMCID: PMC8141241
- DOI: 10.1186/s40168-020-00957-z
Meanders as a scaling motif for understanding of floodplain soil microbiome and biogeochemical potential at the watershed scale
Abstract
Background: Biogeochemical exports from watersheds are modulated by the activity of microorganisms that function over micron scales. Here, we tested the hypothesis that meander-bound regions share a core microbiome and exhibit patterns of metabolic potential that broadly predict biogeochemical processes in floodplain soils along a river corridor.
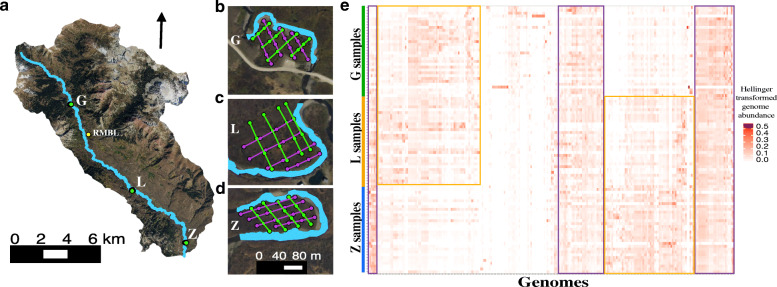
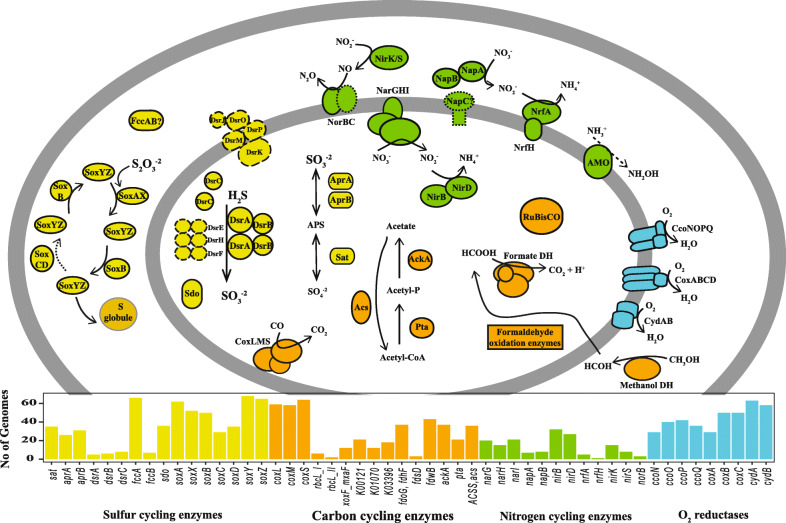
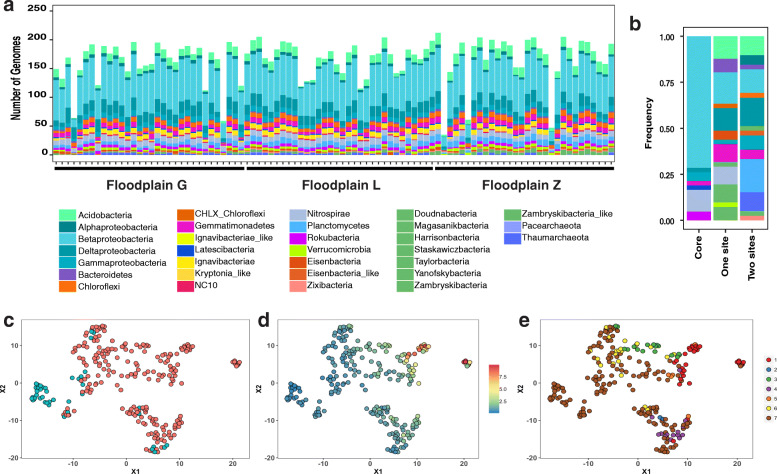
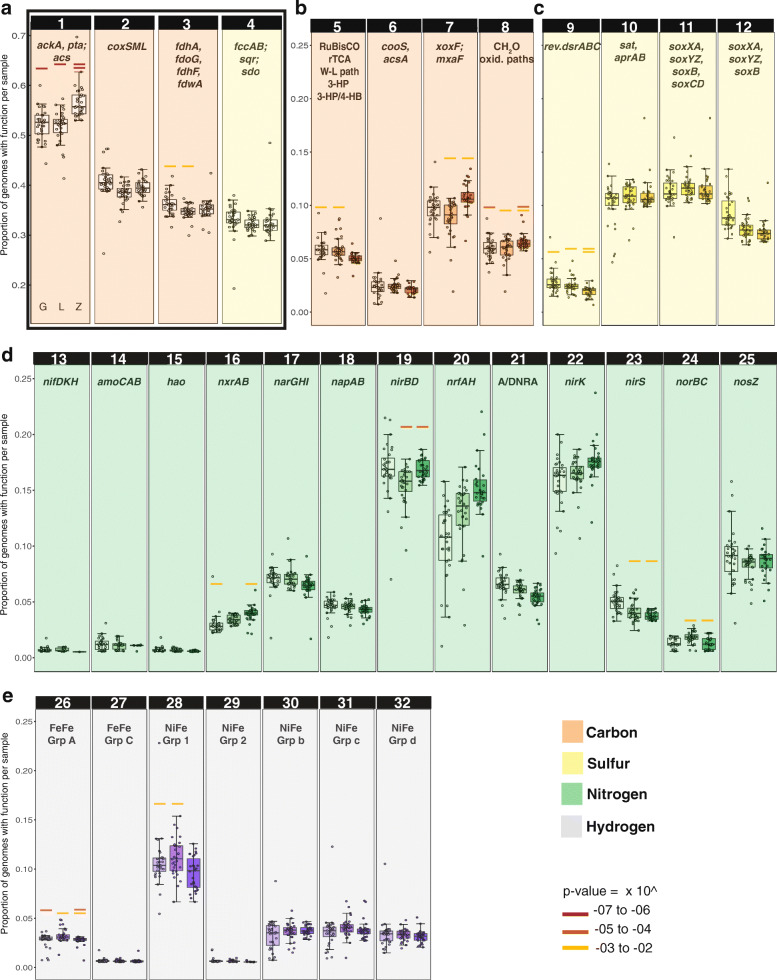
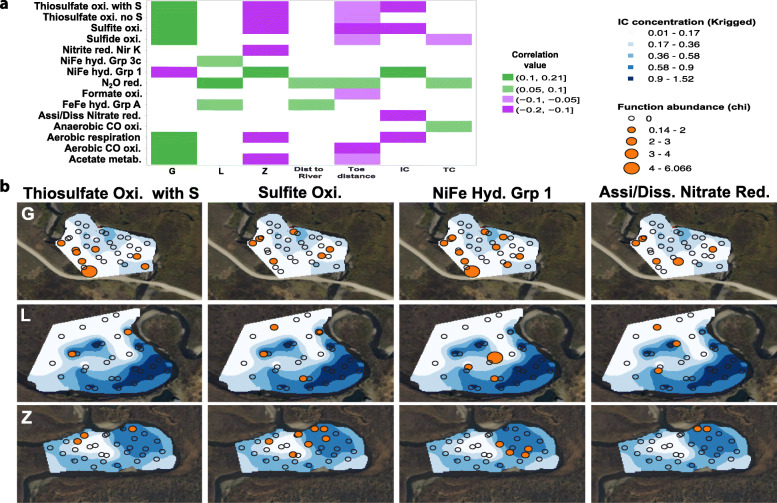
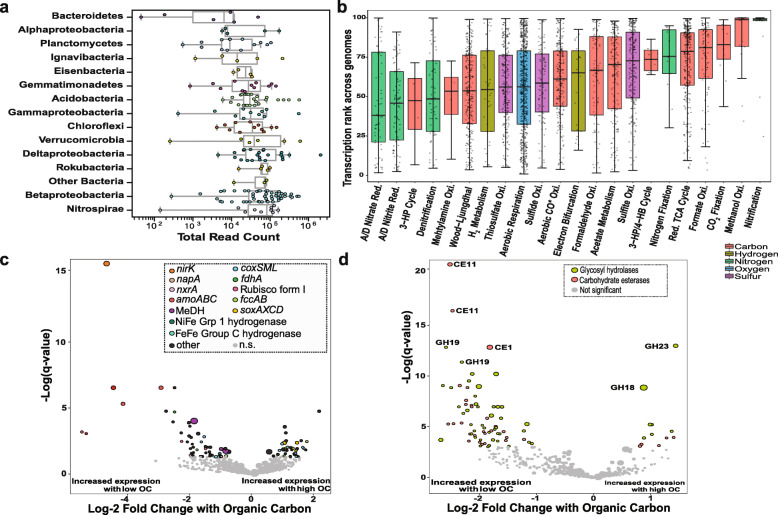
Results: We intensively sampled the microbiomes of floodplain soils located in the upper, middle, and lower reaches of the East River, Colorado. Despite the very high microbial diversity and complexity of the soils, we reconstructed 248 quality draft genomes representative of subspecies. Approximately one third of these bacterial subspecies was detected across all three locations at similar abundance levels, and ~ 15% of species were detected in two consecutive years. Within the meander-bound floodplains, we did not detect systematic patterns of gene abundance based on sampling position relative to the river. However, across meanders, we identified a core floodplain microbiome that is enriched in capacities for aerobic respiration, aerobic CO oxidation, and thiosulfate oxidation with the formation of elemental sulfur. Given this, we conducted a transcriptomic analysis of the middle floodplain. In contrast to predictions made based on the prominence of gene inventories, the most highly transcribed genes were relatively rare amoCAB and nxrAB (for nitrification) genes, followed by genes involved in methanol and formate oxidation, and nitrogen and CO2 fixation. Within all three meanders, low soil organic carbon correlated with high activity of genes involved in methanol, formate, sulfide, hydrogen, and ammonia oxidation, nitrite oxidoreduction, and nitrate and nitrite reduction. Overall, the results emphasize the importance of sulfur, one-carbon and nitrogen compound metabolism in soils of the riparian corridor.
Conclusions: The disparity between the scale of a microbial cell and the scale of a watershed currently limits the development of genomically informed predictive models describing watershed biogeochemical function. Meander-bound floodplains appear to serve as scaling motifs that predict aggregate capacities for biogeochemical transformations, providing a foundation for incorporating riparian soil microbiomes in watershed models. Widely represented genetic capacities did not predict in situ activity at one time point, but rather they define a reservoir of biogeochemical potential available as conditions change. Video abstract.
Keywords: Floodplain; Genome-resolved metagenomics; Metatranscriptomics; Microbiome; Soil; Watershed.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Viviroli D, Dürr HH, Messerli B, Meybeck M, Weingartner R. Mountains of the world, water towers for humanity: typology, mapping, and global significance. Water Resour Res. 2007;43(7). 10.1029/2006wr005653.
-
- Immerzeel WW, Lutz AF, Andrade M, Bahl A, Biemans H, Bolch T, Hyde S, Brumby S, Davies BJ, Elmore AC, Emmer A, Feng M, Fernández A, Haritashya U, Kargel JS, Koppes M, Kraaijenbrink PDA, Kulkarni AV, Mayewski PA, Nepal S, Pacheco P, Painter TH, Pellicciotti F, Rajaram H, Rupper S, Sinisalo A, Shrestha AB, Viviroli D, Wada Y, Xiao C, Yao T, Baillie JEM. Importance and vulnerability of the world’s water towers. Nature. 2020;577(7790):364–369. doi: 10.1038/s41586-019-1822-y. - DOI - PubMed
-
- Hubbard SS, Williams KH, Agarwal D, Banfield J, Beller H, Bouskill N, Brodie E, Carroll R, Dafflon B, Dwivedi D, Falco N, Faybishenko B, Maxwell R, Nico P, Steefel C, Steltzer H, Tokunaga T, Tran PA, Wainwright H, Varadharajan C. The East River, Colorado, watershed: a mountainous community testbed for improving predictive understanding of multiscale hydrological–biogeochemical dynamics. Vadose Zone J. 2018;17(1):180061. doi: 10.2136/vzj2018.03.0061. - DOI
-
- Levin SA. The problem of pattern and scale in ecology: the Robert H. MacArthur Award lecture. Ecology. 1992;73(6):1943–1967. doi: 10.2307/1941447. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
