

Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS)
- PMID: 34024024
- PMCID: PMC8571445
- DOI: 10.1007/s40120-021-00258-z
Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS)
Abstract
Introduction: Hereditary transthyretin amyloidosis (ATTRv amyloidosis) is a clinically heterogeneous disease caused by mutations in the transthyretin (TTR) gene. The most common mutation, Val30Met, can manifest as an early- or late-onset disease.
Methods: The Transthyretin Amyloidosis Outcomes Survey (THAOS) is an ongoing, global, longitudinal, observational survey of patients with transthyretin amyloidosis, including both inherited and wild-type disease and asymptomatic patients with TTR mutations. This is a descriptive analysis of symptomatic patients with ATTRv Val30Met amyloidosis with late- (age at least 50 years) vs. early-onset (age less than 50 years) disease in THAOS (data cutoff August 1, 2019).
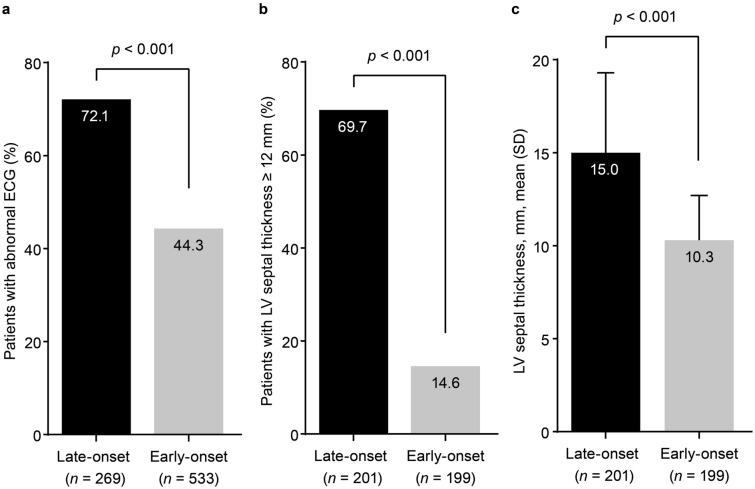
Results: Of 1389 patients with ATTRv Val30Met amyloidosis, 491 (35.3%) had late-onset disease. Compared with early-onset, patients with late-onset were more likely to be male (66.2% vs. 53.6%) and have a longer mean (standard deviation [SD]) time from onset to diagnosis (3.8 [3.4] vs. 2.7 [4.1] years). Late-onset disease was associated with more severe neurological impairment at enrollment (median [10th, 90th percentile] derived Neuropathy Impairment Score in the Lower Limbs, 25.0 [4.0, 69.3] vs. 8.0 [0, 54.8]; Neurologic Composite Score, 42.0 [2.0, 155.0] vs. 21.0 [0, 102.0]). Cardiac findings were more prominent in late-onset disease. An overall interpretation of electrocardiogram as abnormal was reported in 72.1% of late-onset patients (vs. 44.3% early-onset). A left-ventricular septal thickness of at least 12 mm was reported in 69.7% of late-onset patients (vs. 14.6% early-onset). All differences were statistically significant (p < 0.001).
Conclusion: In THAOS, late-onset ATTRv Val30Met amyloidosis is common, presenting with more severe neurologic and cardiac findings at enrollment. Heterogeneity of disease may make it more difficult to diagnose. Increased recognition of late-onset ATTRv Val30Met amyloidosis could lead to more timely diagnosis and improve patient outcomes.
Trial registration: ClinicalTrials.gov NCT00628745.
Keywords: ATTRv amyloidosis; Cardiac; Disease onset; Neurologic.
© 2021. The Author(s).
Figures

References
-
- Rowczenio D, Wechalekar A. Mutations in hereditary amyloidosis. http://amyloidosismutations.com/mut-attr.php. 2015. Accessed Dec 18, 2020.
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
