

DNF: A differential network flow method to identify rewiring drivers for gene regulatory networks
- PMID: 34025035
- PMCID: PMC8139126
- DOI: 10.1016/j.neucom.2020.05.028
DNF: A differential network flow method to identify rewiring drivers for gene regulatory networks
Abstract
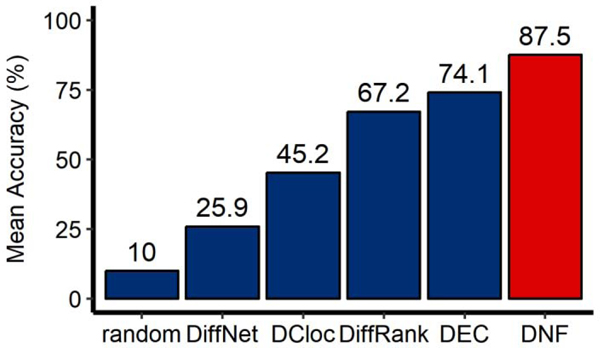
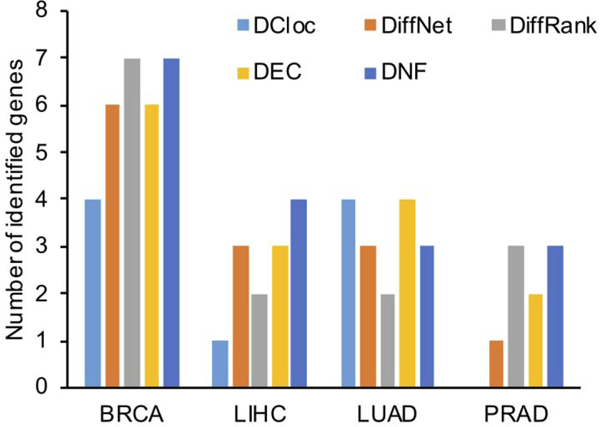
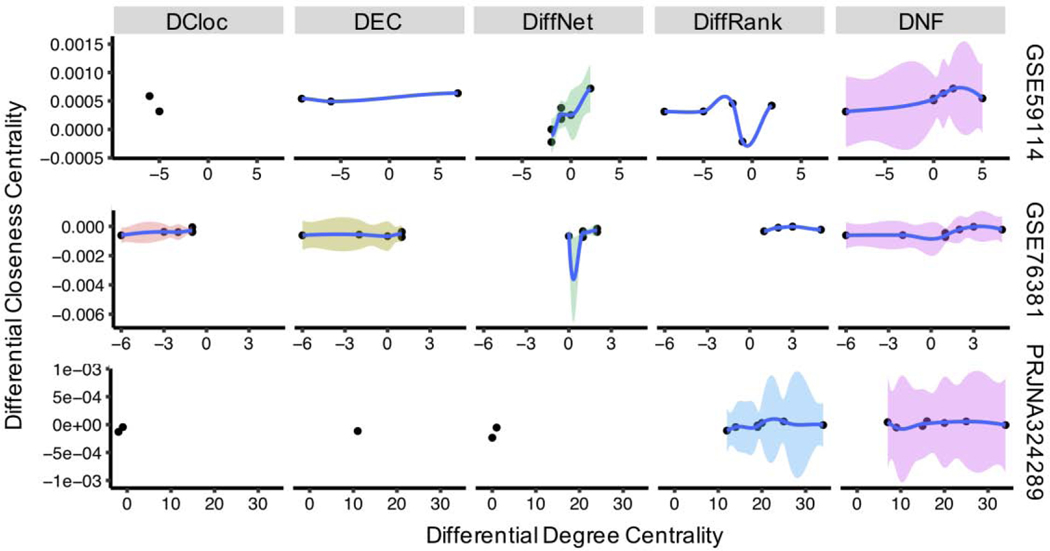
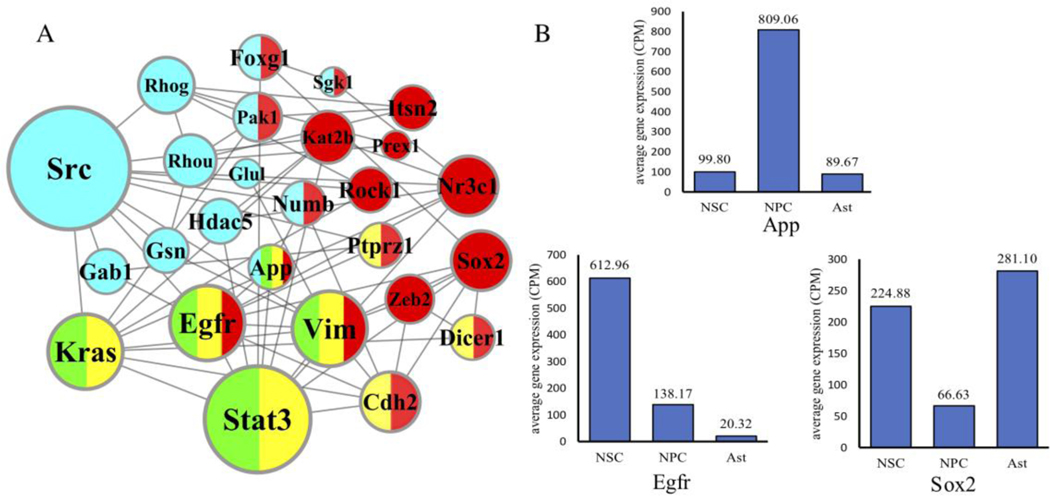
Differential network analysis has become an important approach in identifying driver genes in development and disease. However, most studies capture only local features of the underlying gene-regulatory network topology. These approaches are vulnerable to noise and other changes which mask driver-gene activity. Therefore, methods are urgently needed which can separate the impact of true regulatory elements from stochastic changes and downstream effects. We propose the differential network flow (DNF) method to identify key regulators of progression in development or disease. Given the network representation of consecutive biological states, DNF quantifies the essentiality of each node by differences in the distribution of network flow, which are capable of capturing comprehensive topological differences from local to global feature domains. DNF achieves more accurate driver-gene identification than other state-of-the-art methods when applied to four human datasets from The Cancer Genome Atlas and three single-cell RNA-seq datasets of murine neural and hematopoietic differentiation. Furthermore, we predict key regulators of crosstalk between separate networks underlying both neuronal differentiation and the progression of neurodegenerative disease, among which APP is predicted as a driver gene of neural stem cell differentiation. Our method is a new approach for quantifying the essentiality of genes across networks of different biological states.
Keywords: differential network analysis; information entropy; network flow; network topology; neuronal differentiation.
Figures
References
-
- Barzel B, Liu YY, Barabasi AL, Constructing minimal models for complex system dynamics, Nature Communications, 6 (2015) 7186. - PubMed