

Cellular Mechanisms of Liver Fibrosis
- PMID: 34025430
- PMCID: PMC8134740
- DOI: 10.3389/fphar.2021.671640
Cellular Mechanisms of Liver Fibrosis
Abstract
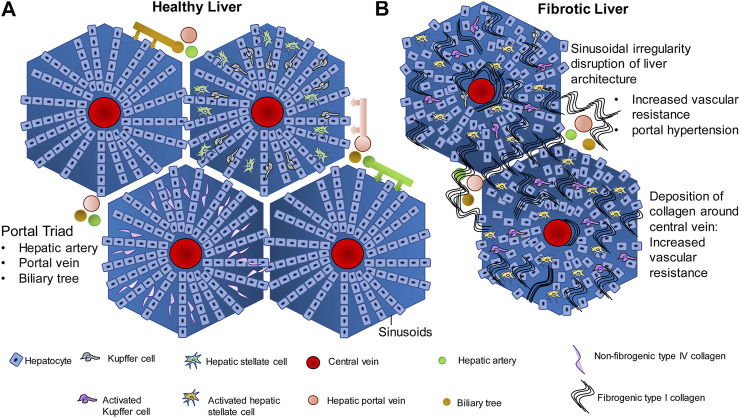
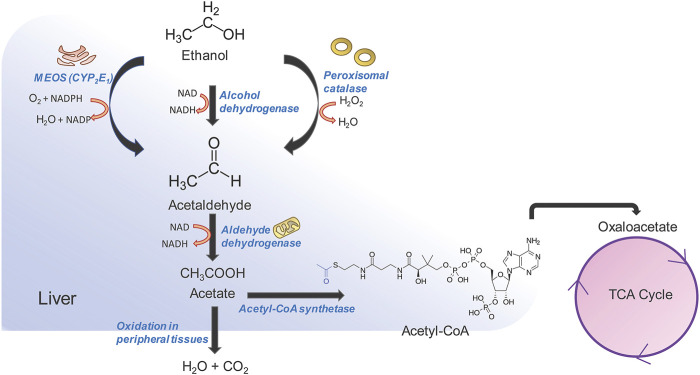
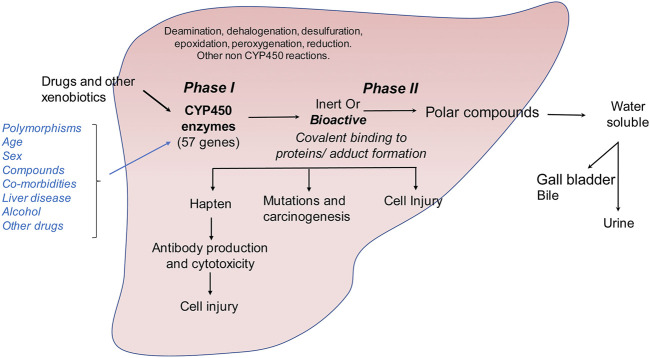
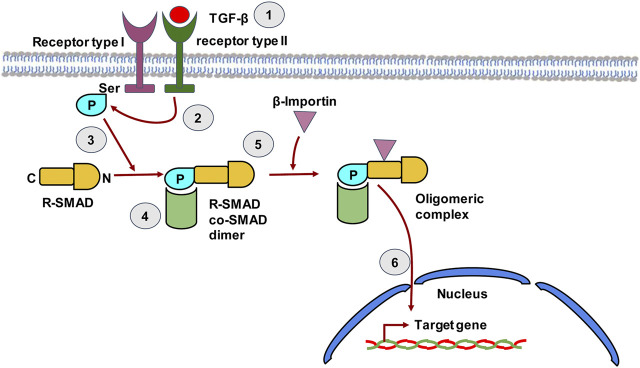
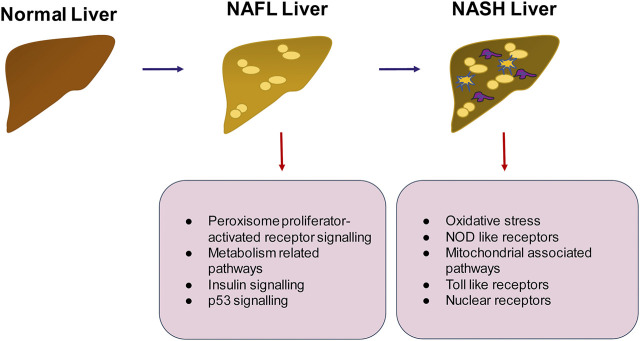
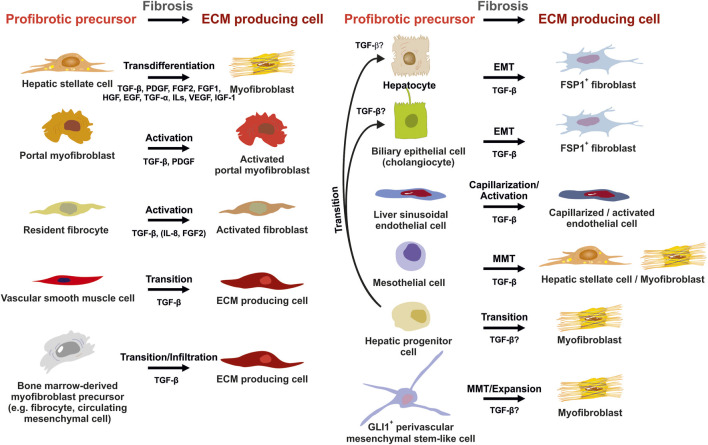
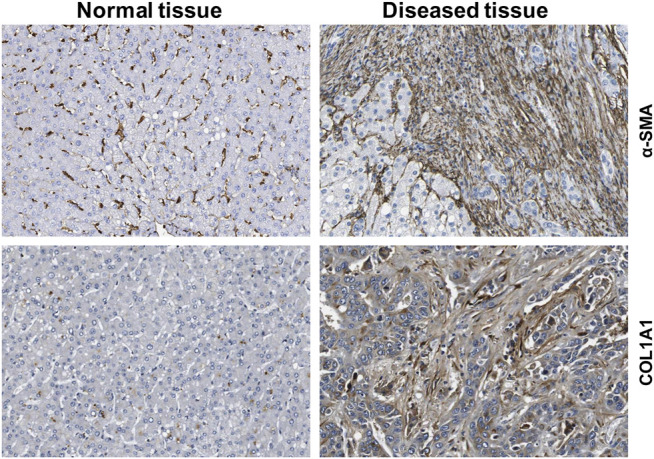
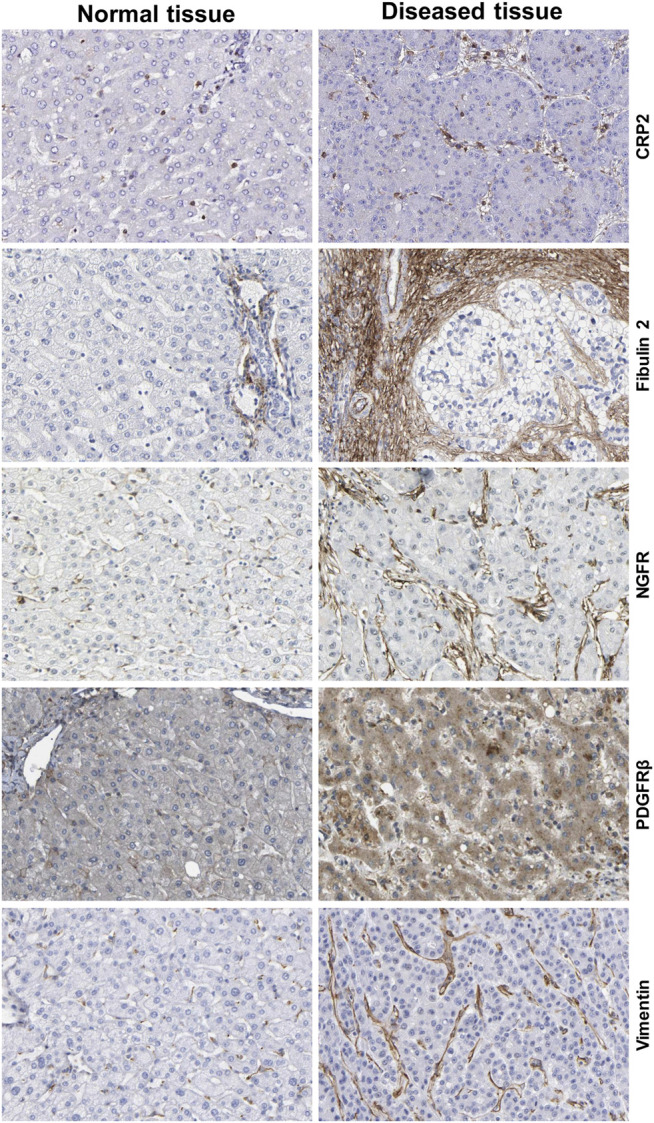
The liver is a central organ in the human body, coordinating several key metabolic roles. The structure of the liver which consists of the distinctive arrangement of hepatocytes, hepatic sinusoids, the hepatic artery, portal vein and the central vein, is critical for its function. Due to its unique position in the human body, the liver interacts with components of circulation targeted for the rest of the body and in the process, it is exposed to a vast array of external agents such as dietary metabolites and compounds absorbed through the intestine, including alcohol and drugs, as well as pathogens. Some of these agents may result in injury to the cellular components of liver leading to the activation of the natural wound healing response of the body or fibrogenesis. Long-term injury to liver cells and consistent activation of the fibrogenic response can lead to liver fibrosis such as that seen in chronic alcoholics or clinically obese individuals. Unidentified fibrosis can evolve into more severe consequences over a period of time such as cirrhosis and hepatocellular carcinoma. It is well recognized now that in addition to external agents, genetic predisposition also plays a role in the development of liver fibrosis. An improved understanding of the cellular pathways of fibrosis can illuminate our understanding of this process, and uncover potential therapeutic targets. Here we summarized recent aspects in the understanding of relevant pathways, cellular and molecular drivers of hepatic fibrosis and discuss how this knowledge impact the therapy of respective disease.
Keywords: NASH; alcohol; chemokines; cholestasis; cytokines; drugs; liver fibrosis; therapy.
Copyright © 2021 Acharya, Chouhan, Weiskirchen and Weiskirchen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
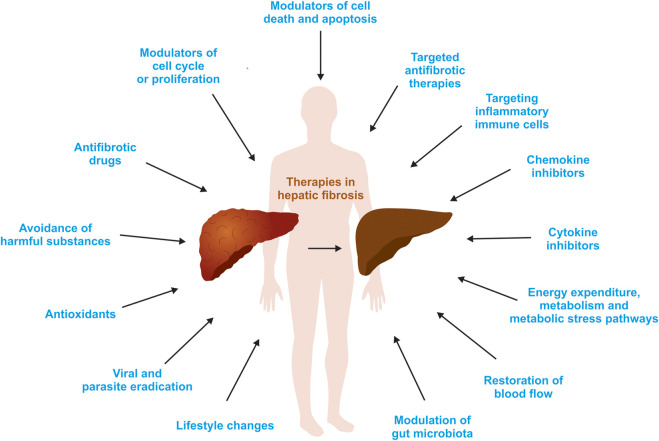
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources