

Genetic predisposition similarities between NASH and ASH: Identification of new therapeutic targets
- PMID: 34027340
- PMCID: PMC8122117
- DOI: 10.1016/j.jhepr.2021.100284
Genetic predisposition similarities between NASH and ASH: Identification of new therapeutic targets
Abstract
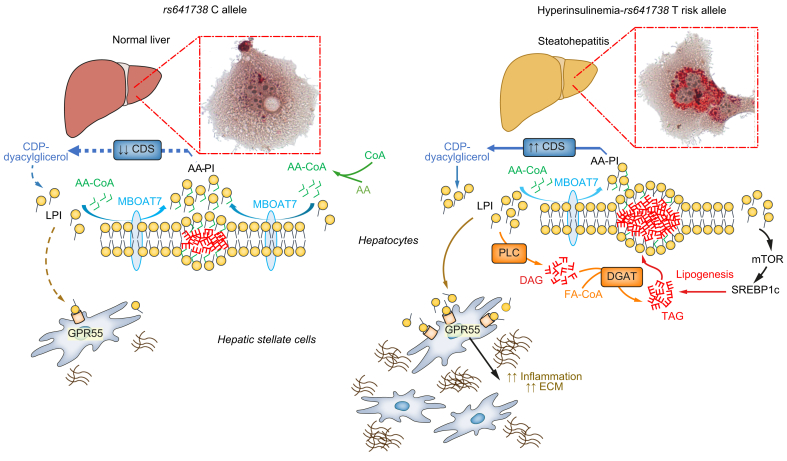
Fatty liver disease can be triggered by a combination of excess alcohol, dysmetabolism and other environmental cues, which can lead to steatohepatitis and can evolve to acute/chronic liver failure and hepatocellular carcinoma, especially in the presence of shared inherited determinants. The recent identification of the genetic causes of steatohepatitis is revealing new avenues for more effective risk stratification. Discovery of the mechanisms underpinning the detrimental effect of causal mutations has led to some breakthroughs in the comprehension of the pathophysiology of steatohepatitis. Thanks to this approach, hepatocellular fat accumulation, altered lipid droplet remodelling and lipotoxicity have now taken centre stage, while the role of adiposity and gut-liver axis alterations have been independently validated. This process could ignite a virtuous research cycle that, starting from human genomics, through omics approaches, molecular genetics and disease models, may lead to the development of new therapeutics targeted to patients at higher risk. Herein, we also review how this knowledge has been applied to: a) the study of the main PNPLA3 I148M risk variant, up to the stage of the first in-human therapeutic trials; b) highlight a role of MBOAT7 downregulation and lysophosphatidyl-inositol in steatohepatitis; c) identify IL-32 as a candidate mediator linking lipotoxicity to inflammation and liver disease. Although this precision medicine drug discovery pipeline is mainly being applied to non-alcoholic steatohepatitis, there is hope that successful products could be repurposed to treat alcohol-related liver disease as well.
Keywords: AA, arachidonic acid; ASH, alcoholic steatohepatitis; DAG, diacylglycerol; DNL, de novo lipogenesis; ER, endoplasmic reticulum; FFAs, free fatty acids; FGF19, fibroblast growth factor 19; FLD, fatty liver disease; FXR, farnesoid X receptor; GCKR, glucokinase regulator; GPR55, G protein-coupled receptor 55; HCC, hepatocellular carcinoma; HFE, homeostatic iron regulator; HSC, hepatic stellate cells; HSD17B13, hydroxysteroid 17-beta dehydrogenase 13; IL-, interleukin-; IL32; LDs, lipid droplets; LPI, lysophosphatidyl-inositol; MARC1, mitochondrial amidoxime reducing component 1; MBOAT7; MBOAT7, membrane bound O-acyltransferase domain-containing 7; NASH, non-alcoholic steatohepatitis; PNPLA3; PNPLA3, patatin like phospholipase domain containing 3; PPAR, peroxisome proliferator-activated receptor; PRS, polygenic risk score; PUFAs, polyunsaturated fatty acids; SREBP, sterol response element binding protein; TAG, triacylglycerol; TNF-α, tumour necrosis factor-α; alcoholic liver disease; cirrhosis; fatty liver disease; genetics; interleukin-32; non-alcoholic fatty liver disease; precision medicine; steatohepatitis; therapy.
© 2021 The Author(s).
Conflict of interest statement
LV has received speaking fees from MSD, Gilead, AlfaSigma and AbbVie, served as a consultant for Gilead, Pfizer, Astra Zeneca, Novo Nordisk, Intercept, Diatech Pharmacogenetics and Ionis Pharmaceuticals, and received research grants from Gilead. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures





Similar articles
-
The ménage à trois of autophagy, lipid droplets and liver disease.Autophagy. 2022 Jan;18(1):50-72. doi: 10.1080/15548627.2021.1895658. Epub 2021 Apr 2. Autophagy. 2022. PMID: 33794741 Free PMC article. Review.
-
Combined alcoholic and non-alcoholic steatohepatitis.JHEP Rep. 2020 May 22;2(3):100101. doi: 10.1016/j.jhepr.2020.100101. eCollection 2020 Jun. JHEP Rep. 2020. PMID: 32514497 Free PMC article. Review.
-
Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis.Aliment Pharmacol Ther. 2020 Jun;51(12):1305-1320. doi: 10.1111/apt.15738. Epub 2020 May 7. Aliment Pharmacol Ther. 2020. PMID: 32383295 Free PMC article. Review.
-
n-3 Polyunsaturated fatty acids for the management of alcoholic liver disease: A critical review.Crit Rev Food Sci Nutr. 2019;59(sup1):S116-S129. doi: 10.1080/10408398.2018.1544542. Epub 2018 Dec 22. Crit Rev Food Sci Nutr. 2019. PMID: 30580553 Review.
-
The Combination of Blueberry Juice and Probiotics Ameliorate Non-Alcoholic Steatohepatitis (NASH) by Affecting SREBP-1c/PNPLA-3 Pathway via PPAR-α.Nutrients. 2017 Feb 27;9(3):198. doi: 10.3390/nu9030198. Nutrients. 2017. PMID: 28264426 Free PMC article.
Cited by
-
MBOAT7 rs641738 Variant Is Not Associated with an Increased Risk of Hepatocellular Carcinoma in a Latin American Cohort.Dig Dis Sci. 2023 Nov;68(11):4212-4220. doi: 10.1007/s10620-023-08104-y. Epub 2023 Sep 8. Dig Dis Sci. 2023. PMID: 37684433 Free PMC article.
-
Advanced 3D bioprinted liver models with human-induced hepatocytes for personalized toxicity screening.J Tissue Eng. 2025 Jan 17;16:20417314241313341. doi: 10.1177/20417314241313341. eCollection 2025 Jan-Dec. J Tissue Eng. 2025. PMID: 39839984 Free PMC article.
-
Top level research in hepatology: COVID-19 and beyond.JHEP Rep. 2021 Jun;3(3):100302. doi: 10.1016/j.jhepr.2021.100302. Epub 2021 May 4. JHEP Rep. 2021. PMID: 33969283 Free PMC article. No abstract available.
-
The intersection between alcohol-related liver disease and nonalcoholic fatty liver disease.Nat Rev Gastroenterol Hepatol. 2023 Dec;20(12):764-783. doi: 10.1038/s41575-023-00822-y. Epub 2023 Aug 15. Nat Rev Gastroenterol Hepatol. 2023. PMID: 37582985 Review.
-
Partitioned polygenic risk scores identify distinct types of metabolic dysfunction-associated steatotic liver disease.Nat Med. 2024 Dec;30(12):3614-3623. doi: 10.1038/s41591-024-03284-0. Epub 2024 Dec 9. Nat Med. 2024. PMID: 39653778 Free PMC article.
References
-
- Eslam M., Sanyal A.J., George J., International Consensus P. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158:1999–2014. e1991. - PubMed
-
- Valenti L., Pelusi S. Redefining fatty liver disease classification in 2020. Liver Int. 2020;40:1016–1017. - PubMed
-
- Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L., Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence and outcomes. Hepatology. 2016;64:73–84. - PubMed
-
- Estes C., Anstee Q.M., Arias-Loste M.T., Bantel H., Bellentani S., Caballeria J. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom and United States for the period 2016–2030. J Hepatol. 2018;69:896–904. - PubMed
-
- Goldberg D., Ditah I.C., Saeian K., Lalehzari M., Aronsohn A., Gorospe E.C. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152:1090–1099. e1091. - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
