

Integrin α9 regulates smooth muscle cell phenotype switching and vascular remodeling
- PMID: 34027892
- PMCID: PMC8262341
- DOI: 10.1172/jci.insight.147134
Integrin α9 regulates smooth muscle cell phenotype switching and vascular remodeling
Abstract
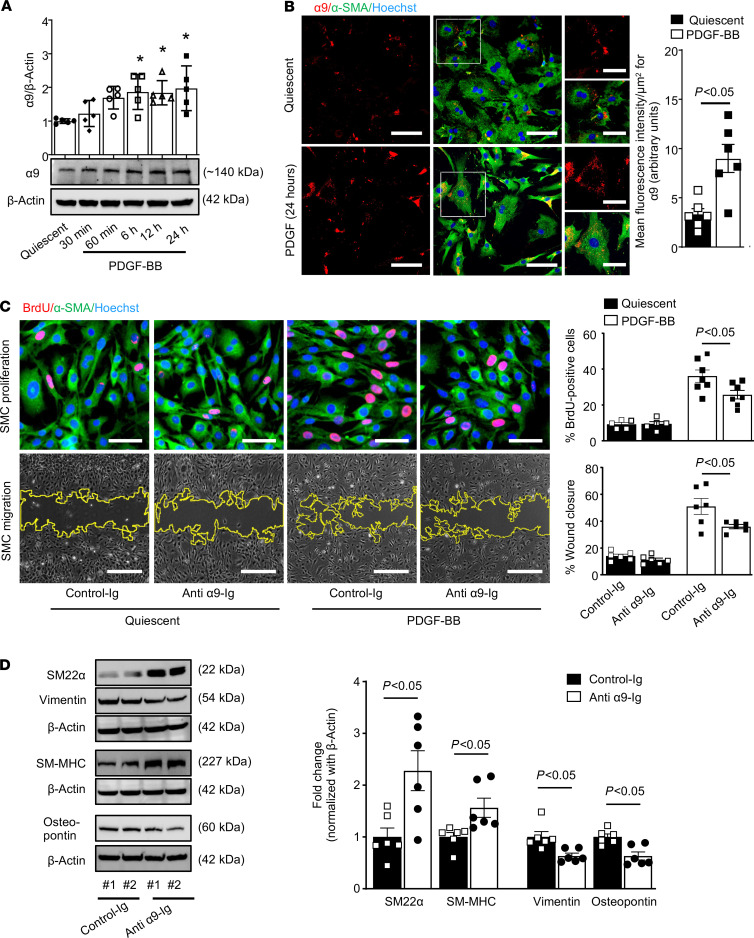
Excessive proliferation of vascular smooth muscle cells (SMCs) remains a significant cause of in-stent restenosis. Integrins, which are heterodimeric transmembrane receptors, play a crucial role in SMC biology by binding to the extracellular matrix protein with the actin cytoskeleton within the SMC. Integrin α9 plays an important role in cell motility and autoimmune diseases; however, its role in SMC biology and remodeling remains unclear. Herein, we demonstrate that stimulated human coronary SMCs upregulate α9 expression. Targeting α9 in stimulated human coronary SMCs, using anti-integrin α9 antibody, suppresses synthetic phenotype and inhibits SMC proliferation and migration. To provide definitive evidence, we generated an SMC-specific α9-deficient mouse strain. Genetic ablation of α9 in SMCs suppressed synthetic phenotype and reduced proliferation and migration in vitro. Mechanistically, suppressed synthetic phenotype and reduced proliferation were associated with decreased focal adhesion kinase/steroid receptor coactivator signaling and downstream targets, including phosphorylated ERK, p38 MAPK, glycogen synthase kinase 3β, and nuclear β-catenin, with reduced transcriptional activation of β-catenin target genes. Following vascular injury, SMC-specific α9-deficient mice or wild-type mice treated with murine anti-integrin α9 antibody exhibited reduced injury-induced neointimal hyperplasia at day 28 by limiting SMC migration and proliferation. Our findings suggest that integrin α9 regulates SMC biology, suggesting its potential therapeutic application in vascular remodeling.
Keywords: Cell migration/adhesion; Integrins; Mouse models; Vascular Biology.
Conflict of interest statement
Figures






References
-
- Shlofmitz E, et al. Restenosis of drug-eluting stents: a new classification system based on disease mechanism to guide treatment and state-of-the-art review. Circ Cardiovasc Interv. 2019;12(8):e007023. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
