

Scaffold Hopping Transformations Using Auxiliary Restraints for Calculating Accurate Relative Binding Free Energies
- PMID: 34029468
- PMCID: PMC8215533
- DOI: 10.1021/acs.jctc.1c00214
Scaffold Hopping Transformations Using Auxiliary Restraints for Calculating Accurate Relative Binding Free Energies
Abstract
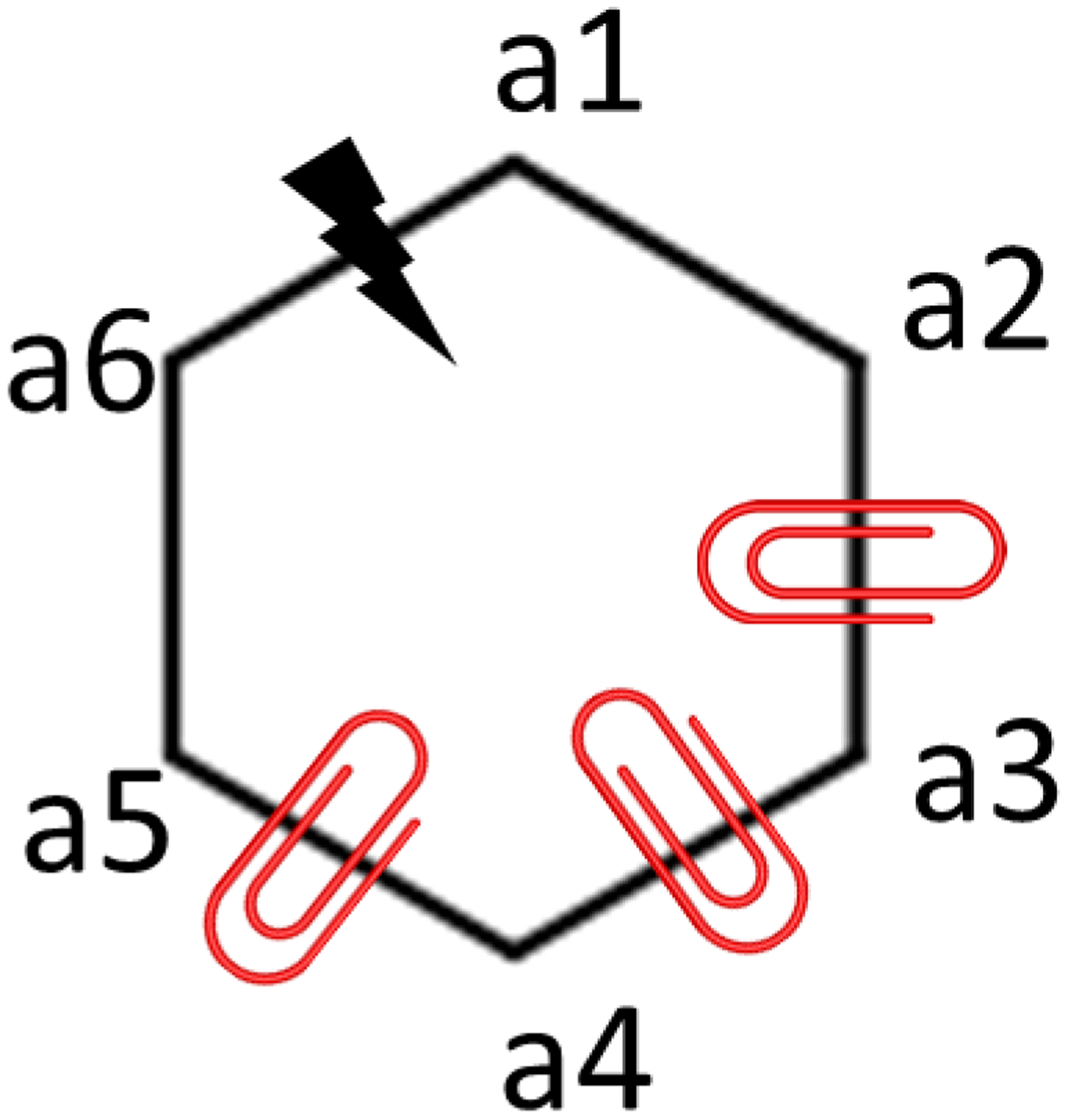
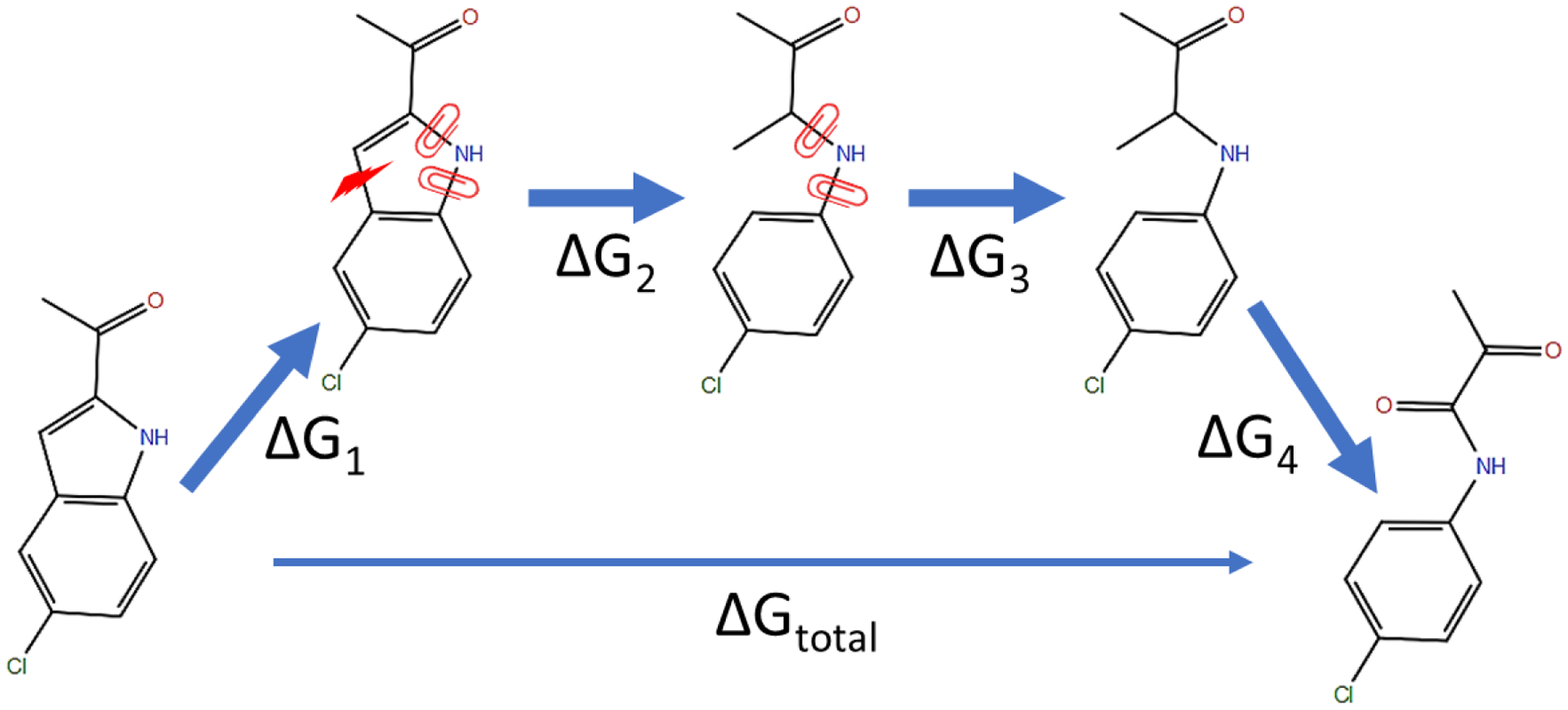
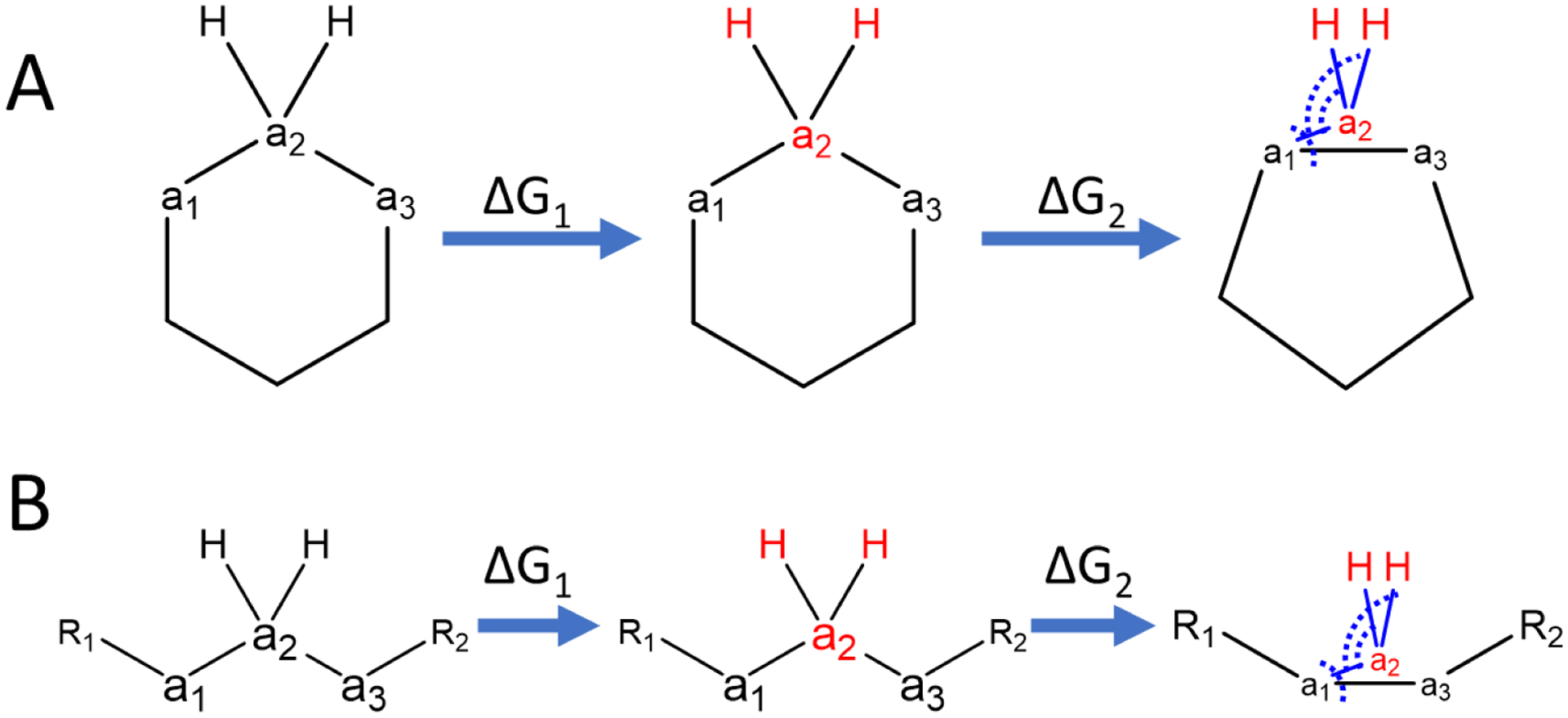
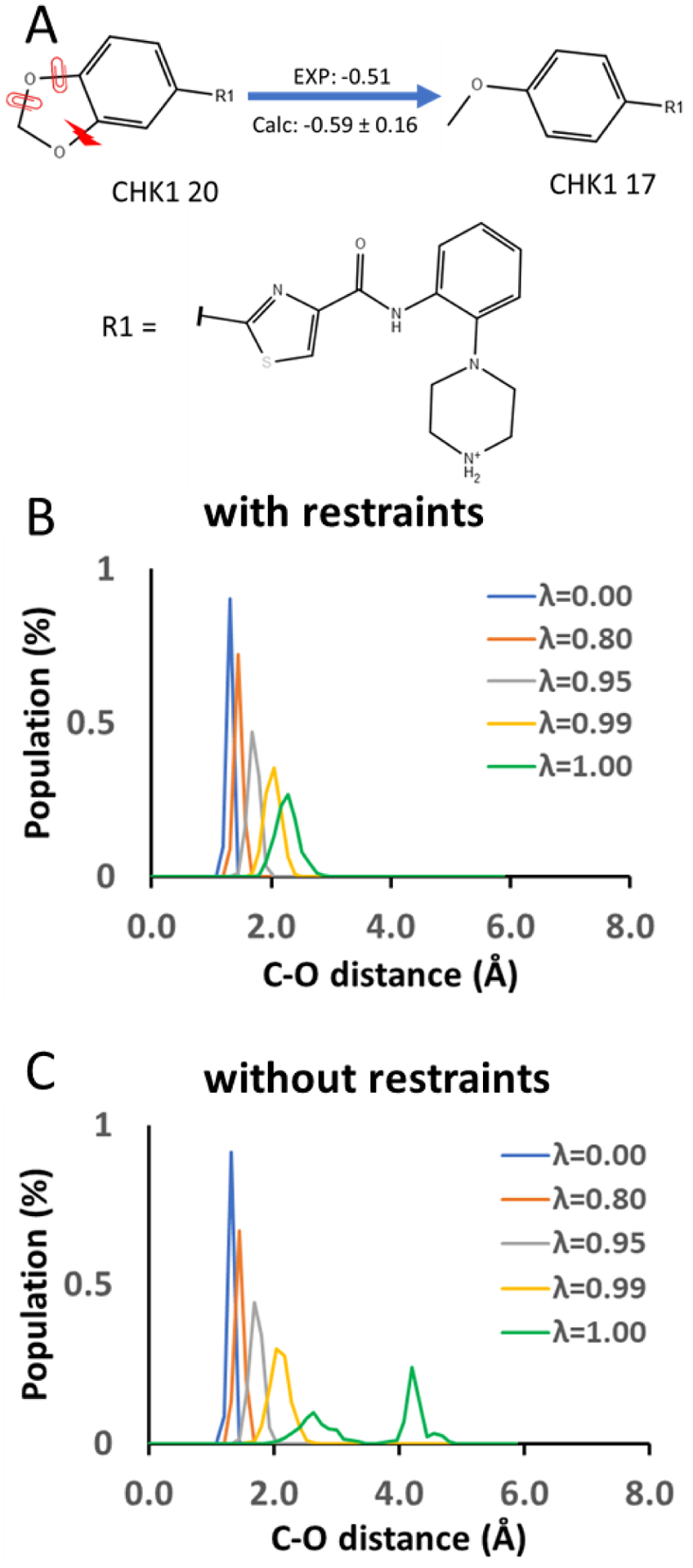
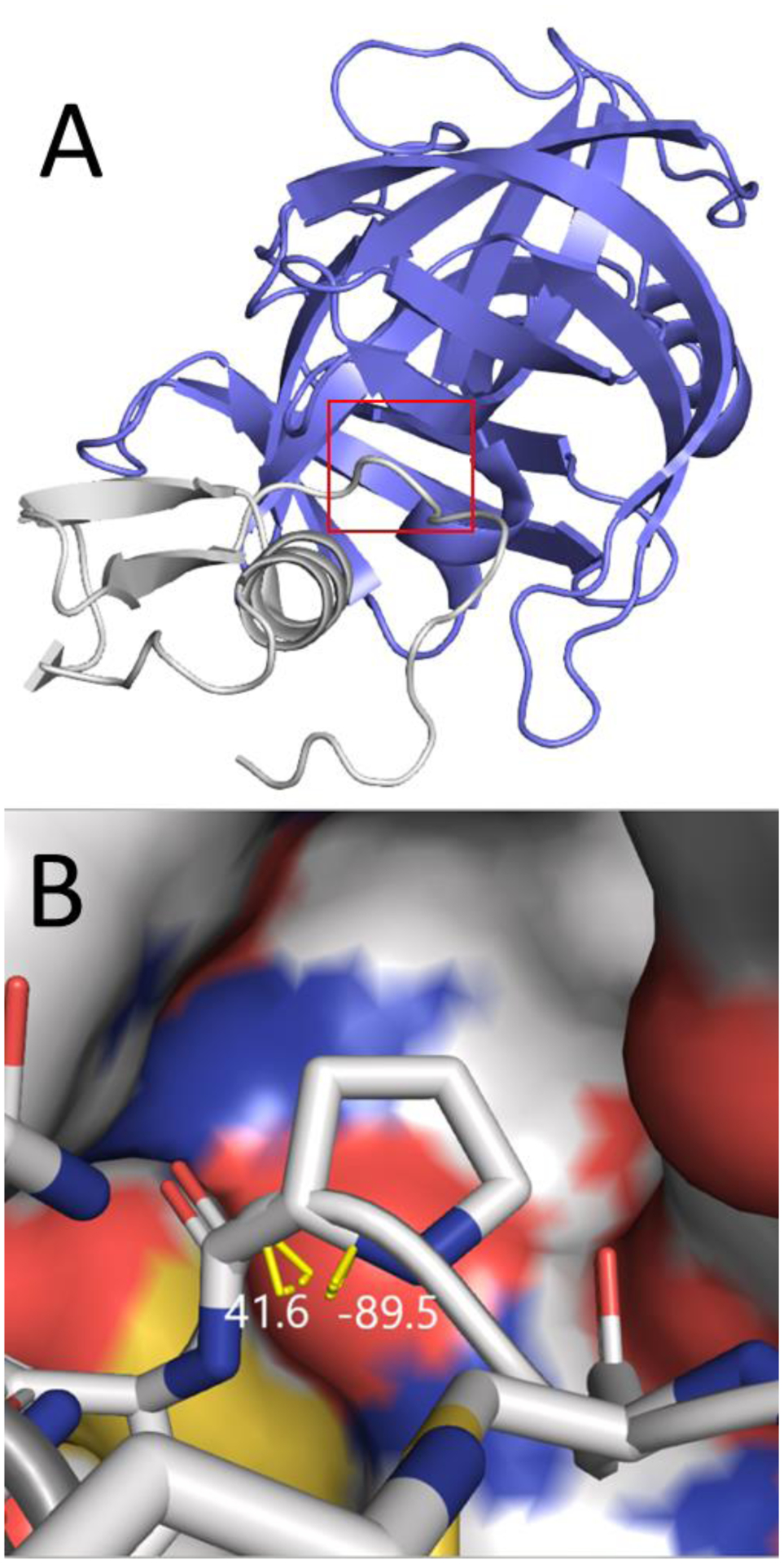
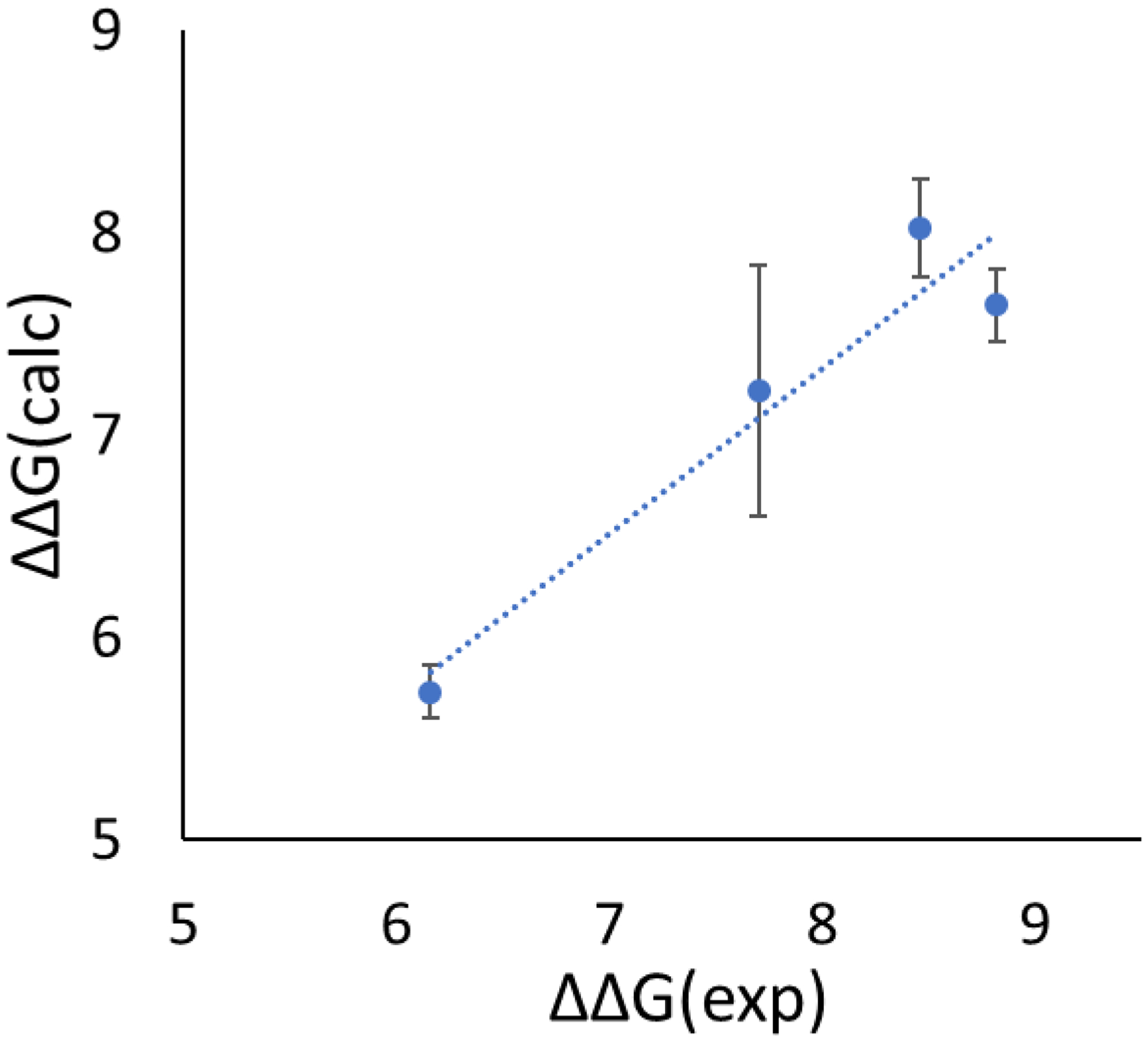
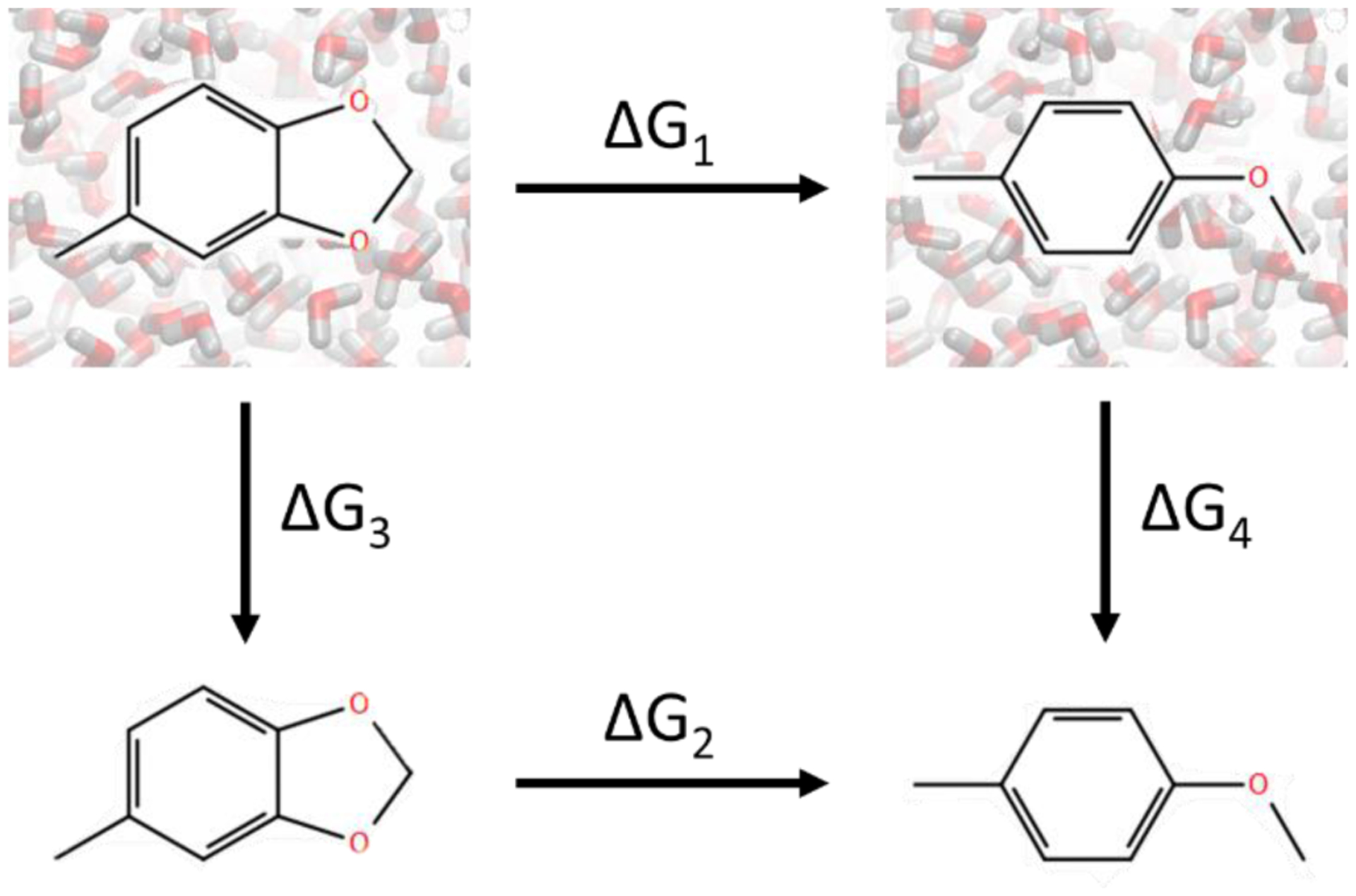
In silico screening of drug-target interactions is a key part of the drug discovery process. Changes in the drug scaffold via contraction or expansion of rings, the breaking of rings, and the introduction of cyclic structures from acyclic structures are commonly applied by medicinal chemists to improve binding affinity and enhance favorable properties of candidate compounds. These processes, commonly referred to as scaffold hopping, are challenging to model computationally. Although relative binding free energy (RBFE) calculations have shown success in predicting binding affinity changes caused by perturbing R-groups attached to a common scaffold, applications of RBFE calculations to modeling scaffold hopping are relatively limited. Scaffold hopping inevitably involves breaking and forming bond interactions of quadratic functional forms, which is highly challenging. A novel method for handling ring opening/closure/contraction/expansion and linker contraction/expansion is presented here. To the best of our knowledge, RBFE calculations on linker contraction/expansion have not been previously reported. The method uses auxiliary restraints to hold the atoms at the ends of a bond in place during the breaking and forming of the bonds. The broad applicability of the method was demonstrated by examining perturbations involving small-molecule macrocycles and mutations of proline in proteins. High accuracy was obtained using the method for most of the perturbations studied. The rigor of the method was isolated from the force field by validating the method using relative and absolute hydration free energy calculations compared to standard simulation results. Unlike other methods that rely on λ-dependent functional forms for bond interactions, the method presented here can be employed using modern molecular dynamics software without modification of codes or force field functions.
Conflict of interest statement
Conflict of interest
JZ, ZL(i), SL, CP, DF, XW, ZL(in) and MY are employees of Xtalpi Inc. TL, CS and DPR declare no conflicts.
Figures
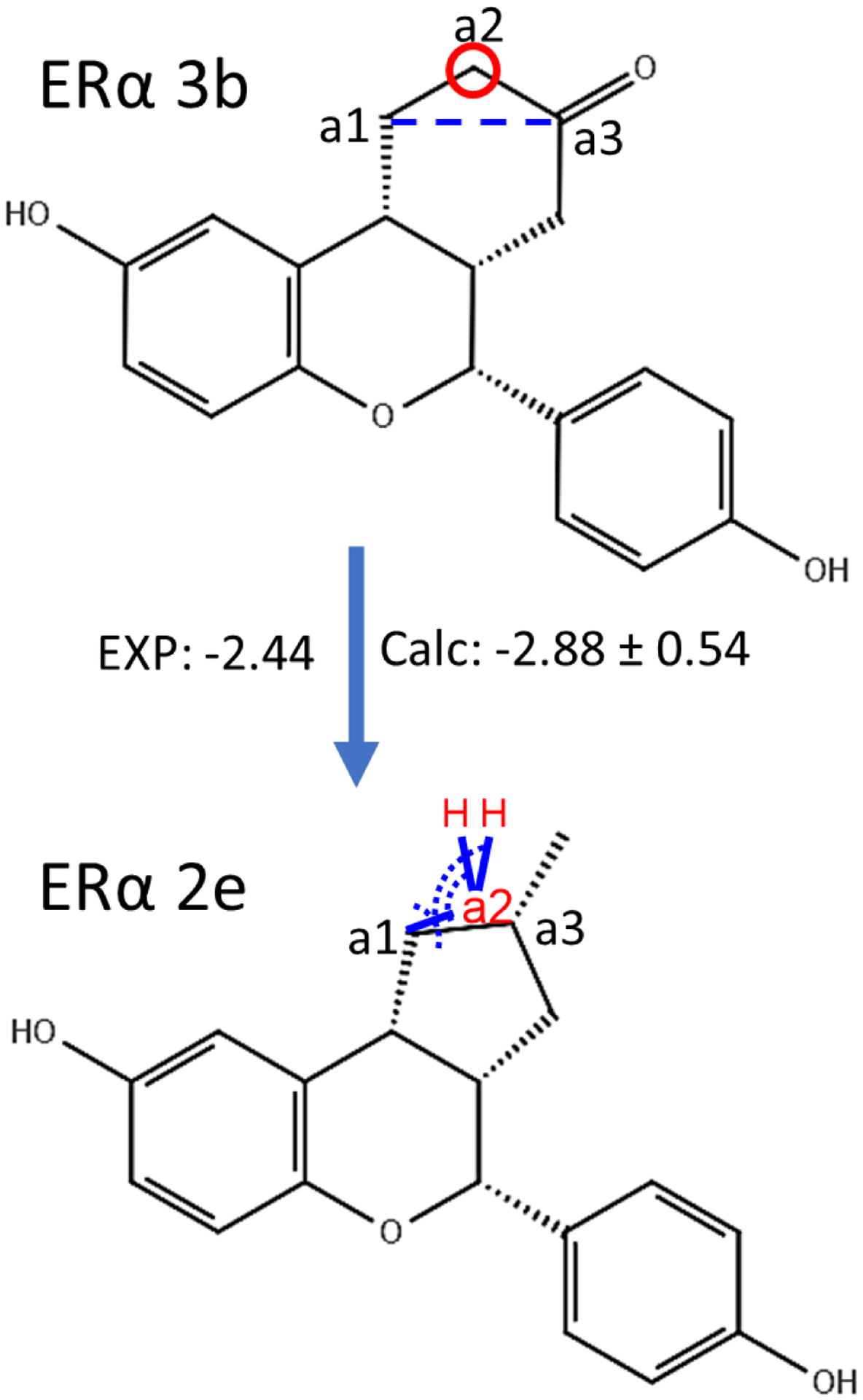
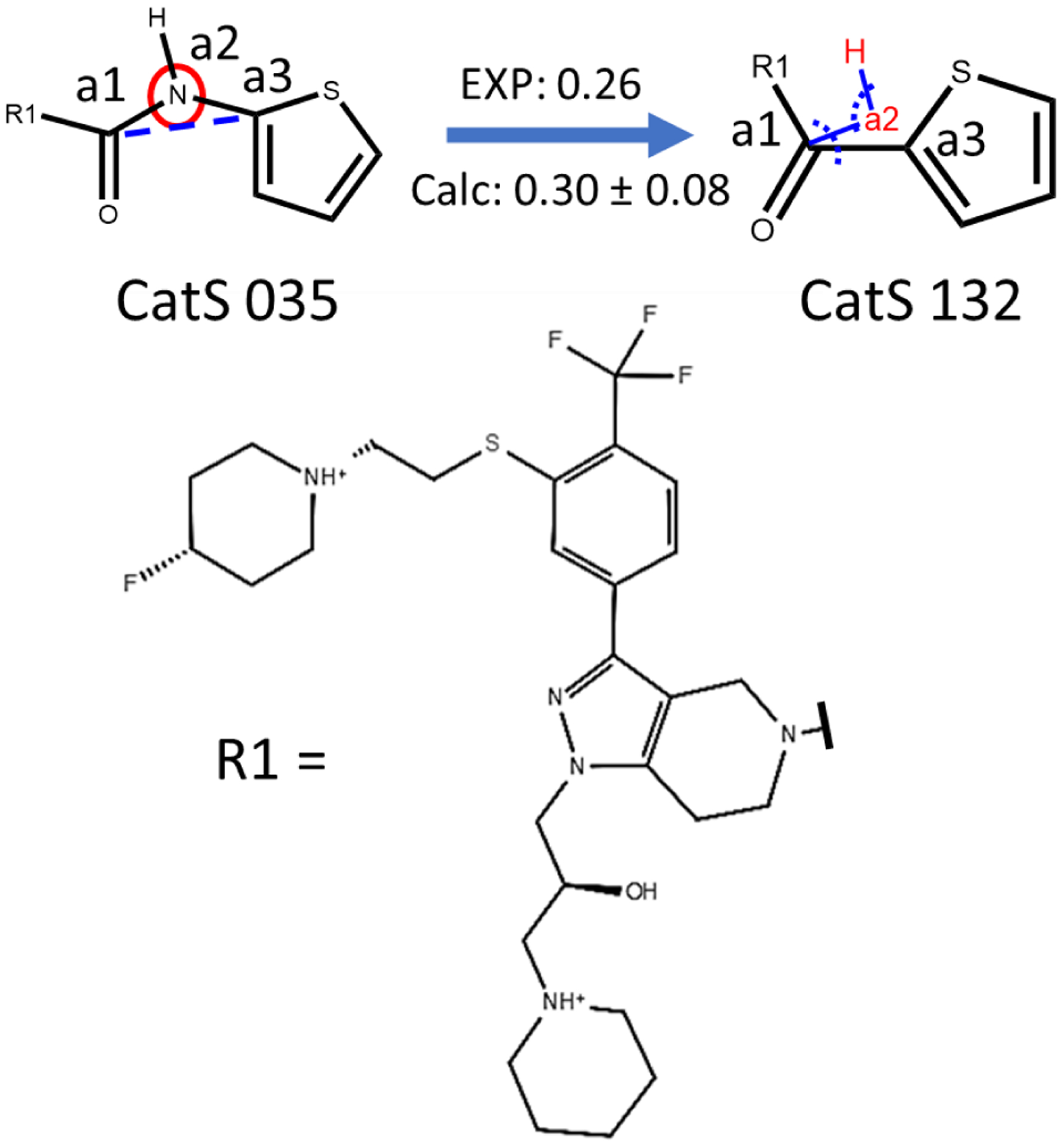
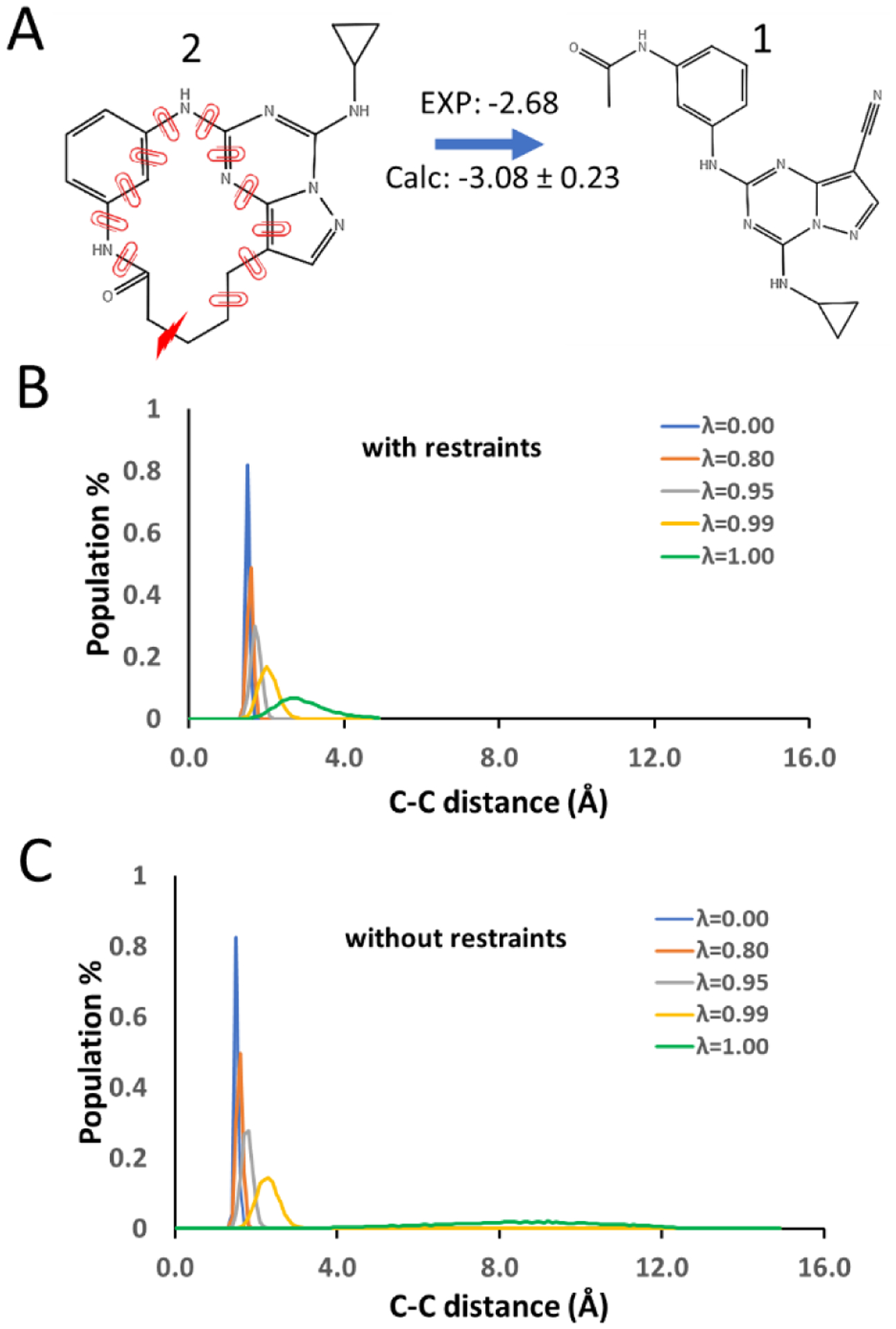
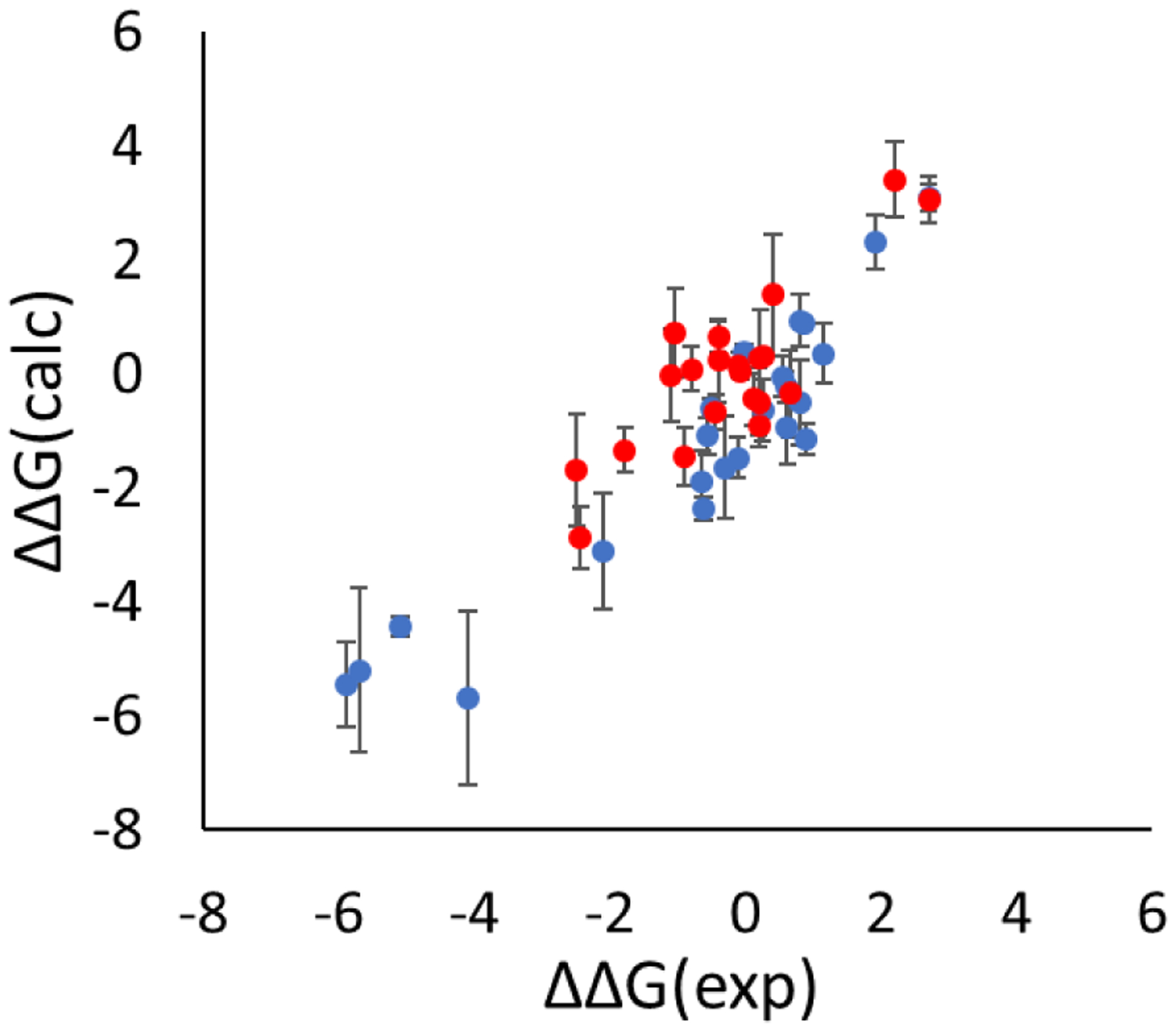
Similar articles
-
Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations.J Chem Inf Model. 2017 Dec 26;57(12):2911-2937. doi: 10.1021/acs.jcim.7b00564. Epub 2017 Dec 15. J Chem Inf Model. 2017. PMID: 29243483
-
Accurate Modeling of Scaffold Hopping Transformations in Drug Discovery.J Chem Theory Comput. 2017 Jan 10;13(1):42-54. doi: 10.1021/acs.jctc.6b00991. Epub 2016 Dec 9. J Chem Theory Comput. 2017. PMID: 27933808
-
Free energy perturbation (FEP)-guided scaffold hopping.Acta Pharm Sin B. 2022 Mar;12(3):1351-1362. doi: 10.1016/j.apsb.2021.09.027. Epub 2021 Sep 30. Acta Pharm Sin B. 2022. PMID: 35530128 Free PMC article.
-
Ring Closure and Ring Opening as Useful Scaffold Hopping Tools in Agrochemistry.J Agric Food Chem. 2023 Nov 29;71(47):18133-18140. doi: 10.1021/acs.jafc.3c01416. Epub 2023 May 24. J Agric Food Chem. 2023. PMID: 37223957 Review.
-
Classification of scaffold-hopping approaches.Drug Discov Today. 2012 Apr;17(7-8):310-24. doi: 10.1016/j.drudis.2011.10.024. Epub 2011 Oct 26. Drug Discov Today. 2012. PMID: 22056715 Free PMC article. Review.
Cited by
-
Broadening the Scope of Binding Free Energy Calculations Using a Separated Topologies Approach.J Chem Theory Comput. 2023 Aug 8;19(15):5058-5076. doi: 10.1021/acs.jctc.3c00282. Epub 2023 Jul 24. J Chem Theory Comput. 2023. PMID: 37487138 Free PMC article.
-
Relative Binding Free Energy between Chemically Distant Compounds Using a Bidirectional Nonequilibrium Approach.J Chem Theory Comput. 2022 Jun 14;18(6):4014-4026. doi: 10.1021/acs.jctc.2c00295. Epub 2022 Jun 1. J Chem Theory Comput. 2022. PMID: 35642423 Free PMC article.
-
A Guide to In Silico Drug Design.Pharmaceutics. 2022 Dec 23;15(1):49. doi: 10.3390/pharmaceutics15010049. Pharmaceutics. 2022. PMID: 36678678 Free PMC article. Review.
-
Relative Binding Free Energy Estimation of Congeneric Ligands and Macromolecular Mutants with the Alchemical Transfer Method with Coordinate Swapping.J Chem Inf Model. 2025 Apr 14;65(7):3706-3714. doi: 10.1021/acs.jcim.5c00207. Epub 2025 Mar 26. J Chem Inf Model. 2025. PMID: 40136079 Free PMC article.
-
Identification of indoles as potential endogenous ligands of ERRγ and their modulation on drug binding.Acta Pharmacol Sin. 2025 Sep;46(9):2574-2582. doi: 10.1038/s41401-025-01550-6. Epub 2025 Apr 8. Acta Pharmacol Sin. 2025. PMID: 40200124
References
-
- Abedini A; Raleigh DP, Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: Is there a critical amyloidogenic domain in human IAPP? J. Mol. Biol 2006, 355 (2), 274–281. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources