

Activation of IGF-1 pathway and suppression of atrophy related genes are involved in Epimedium extract (icariin) promoted C2C12 myotube hypertrophy
- PMID: 34031457
- PMCID: PMC8144409
- DOI: 10.1038/s41598-021-89039-0
Activation of IGF-1 pathway and suppression of atrophy related genes are involved in Epimedium extract (icariin) promoted C2C12 myotube hypertrophy
Abstract
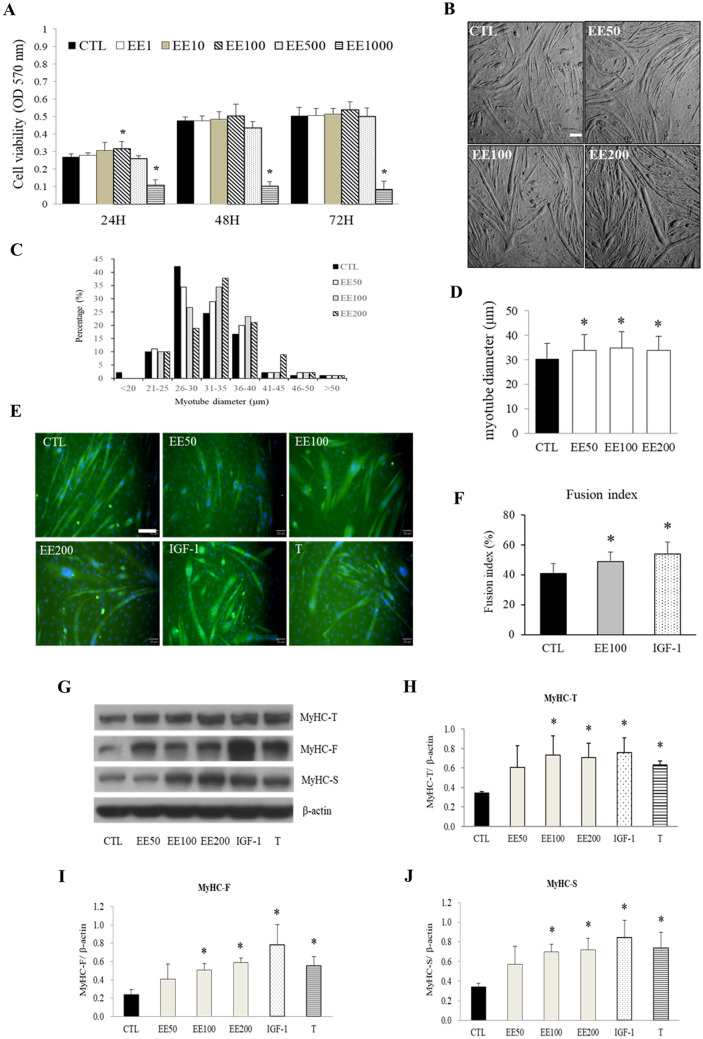
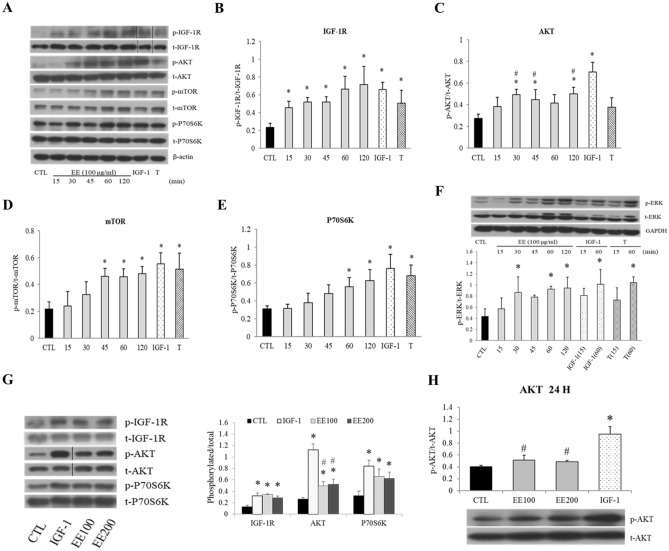
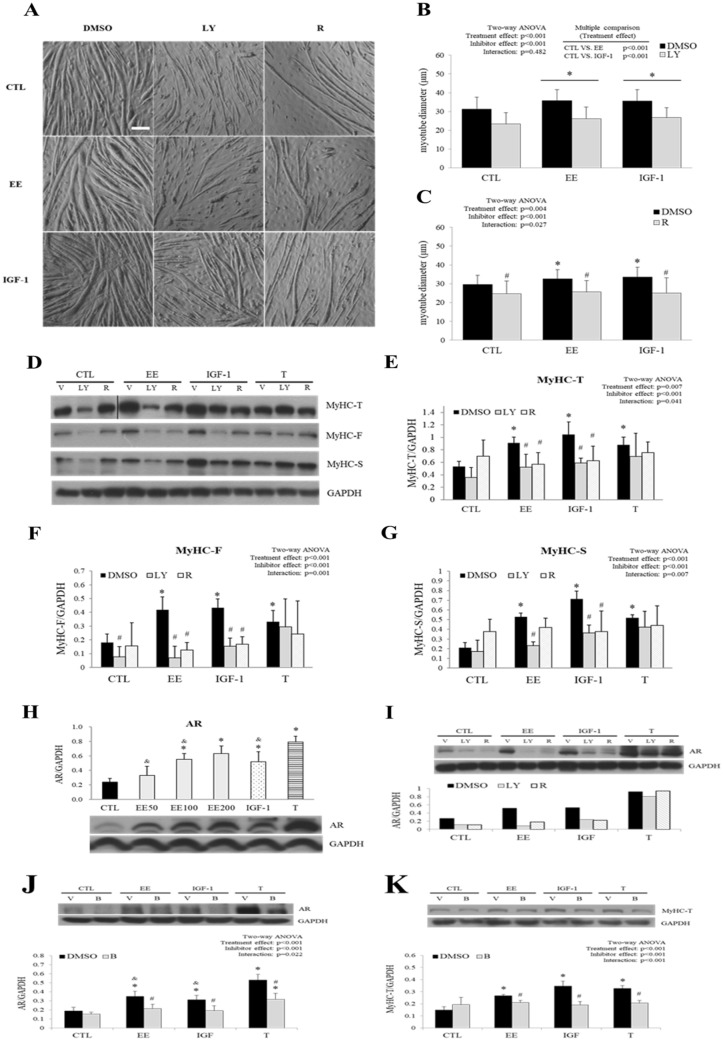
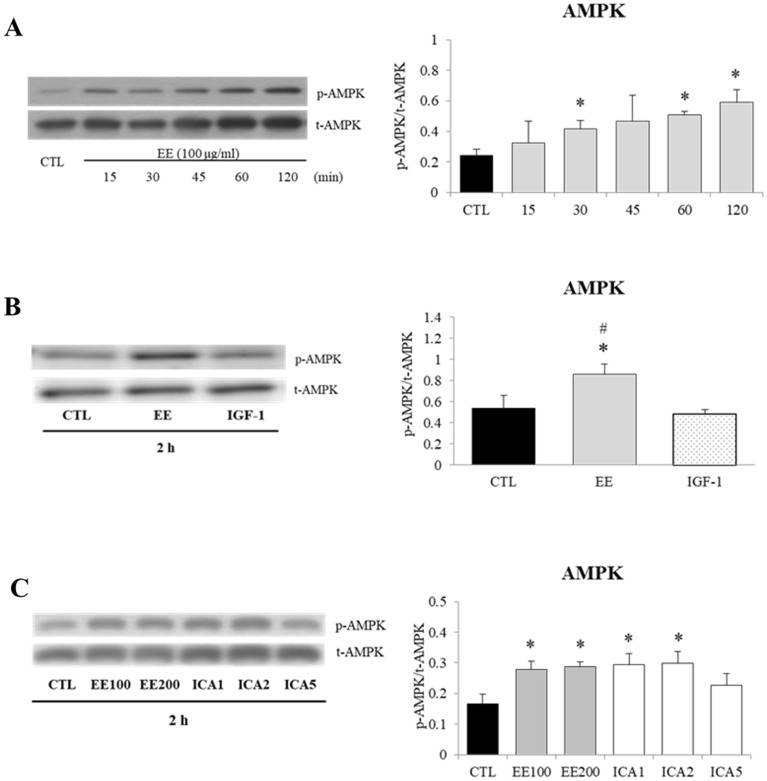
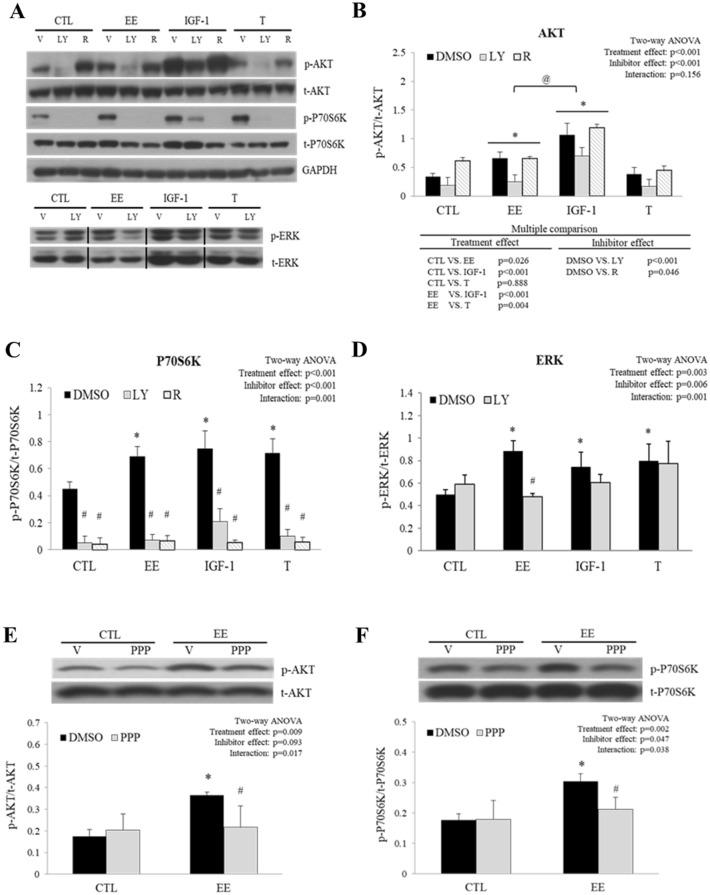
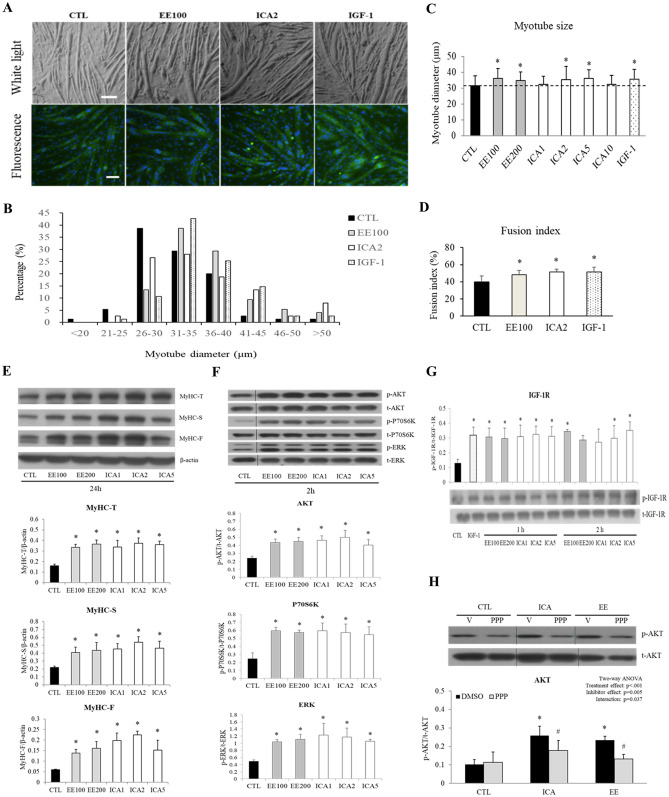
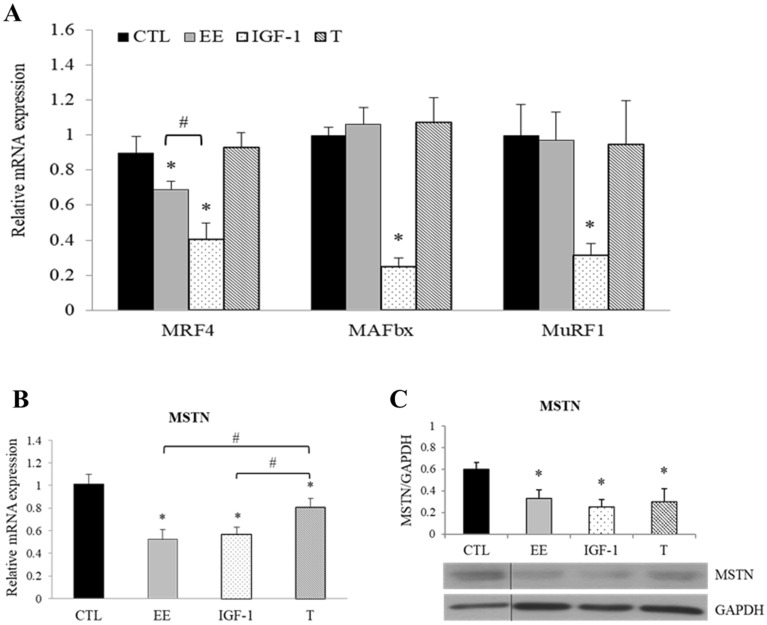
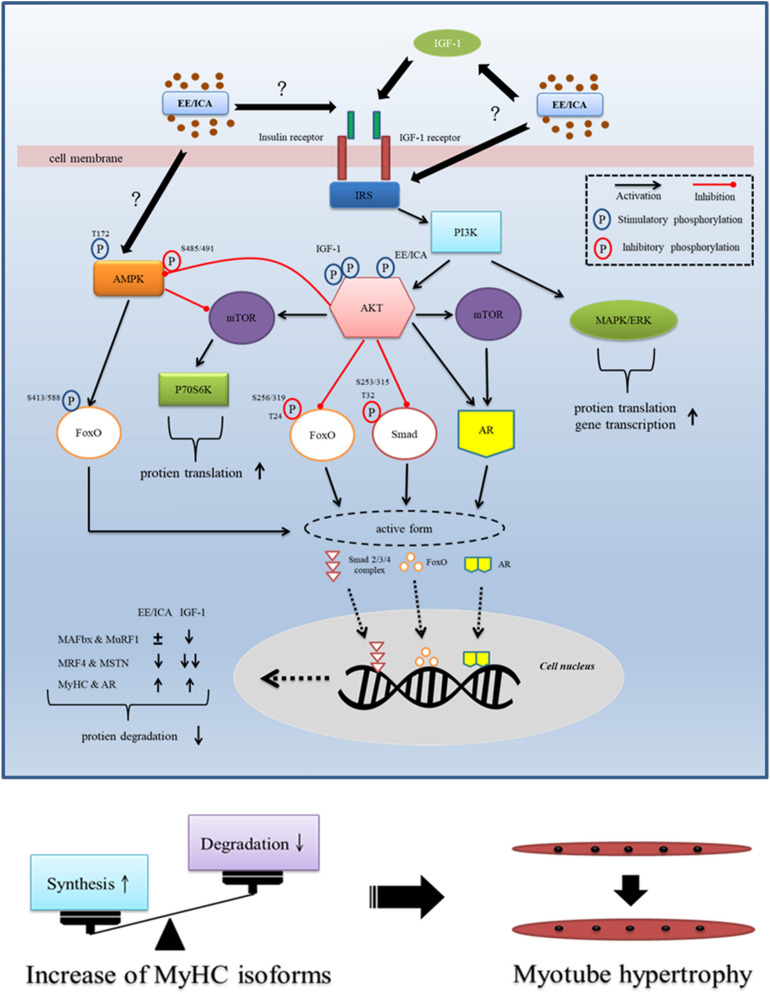
The regenerative effect of Epimedium and its major bioactive flavonoid icariin (ICA) have been documented in traditional medicine, but their effect on sarcopenia has not been evaluated. The aim of this study was to investigate the effects of Epimedium extract (EE) on skeletal muscle as represented by differentiated C2C12 cells. Here we demonstrated that EE and ICA stimulated C2C12 myotube hypertrophy by activating several, including IGF-1 signal pathways. C2C12 myotube hypertrophy was demonstrated by enlarged myotube and increased myosin heavy chains (MyHCs). In similar to IGF-1, EE/ICA activated key components of the IGF-1 signal pathway, including IGF-1 receptor. Pre-treatment with IGF-1 signal pathway specific inhibitors such as picropodophyllin, LY294002, and rapamycin attenuated EE induced myotube hypertrophy and MyHC isoform overexpression. In a different way, EE induced MHyC-S overexpression can be blocked by AMPK, but not by mTOR inhibitor. On the level of transcription, EE suppressed myostatin and MRF4 expression, but did not suppress atrogenes MAFbx and MuRF1 like IGF-1 did. Differential regulation of MyHC isoform and atrogenes is probably due to inequivalent AKT and AMPK phosphorylation induced by EE and IGF-1. These findings suggest that EE/ICA stimulates pathways partially overlapping with IGF-1 signaling pathway to promote myotube hypertrophy.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Muscle-specific overexpression of the type 1 IGF receptor results in myoblast-independent muscle hypertrophy via PI3K, and not calcineurin, signaling.Am J Physiol Endocrinol Metab. 2007 Dec;293(6):E1538-51. doi: 10.1152/ajpendo.00160.2007. Epub 2007 Oct 16. Am J Physiol Endocrinol Metab. 2007. PMID: 17940216
-
Glycogen synthase kinase-3β is required for the induction of skeletal muscle atrophy.Am J Physiol Cell Physiol. 2011 Nov;301(5):C995-C1007. doi: 10.1152/ajpcell.00520.2010. Epub 2011 Aug 10. Am J Physiol Cell Physiol. 2011. PMID: 21832246
-
The collagen derived dipeptide hydroxyprolyl-glycine promotes C2C12 myoblast differentiation and myotube hypertrophy.Biochem Biophys Res Commun. 2016 Sep 23;478(3):1292-7. doi: 10.1016/j.bbrc.2016.08.114. Epub 2016 Aug 21. Biochem Biophys Res Commun. 2016. PMID: 27553280
-
PI3 kinase regulation of skeletal muscle hypertrophy and atrophy.Curr Top Microbiol Immunol. 2010;346:267-78. doi: 10.1007/82_2010_78. Curr Top Microbiol Immunol. 2010. PMID: 20593312 Review.
-
Icariin as an emerging candidate drug for anticancer treatment: Current status and perspective.Biomed Pharmacother. 2023 Jan;157:113991. doi: 10.1016/j.biopha.2022.113991. Epub 2022 Nov 9. Biomed Pharmacother. 2023. PMID: 36370524 Review.
Cited by
-
Mechanism of Qigu capsule as a treatment for sarcopenia based on network pharmacology and experimental validation.J Tradit Chin Med. 2025 Apr;45(2):399-407. doi: 10.19852/j.cnki.jtcm.2025.02.001. J Tradit Chin Med. 2025. PMID: 40151126 Free PMC article.
-
Lean nonalcoholic fatty liver disease and sarcopenia.Front Endocrinol (Lausanne). 2023 Jun 23;14:1217249. doi: 10.3389/fendo.2023.1217249. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 37424859 Free PMC article. Review.
-
A clinical herbal prescription Gu-Shu-Kang capsule exerted beneficial effects on the musculoskeletal system of dexamethasone-treated mice by acting on tissue IGF-1 signalling pathway.Pharm Biol. 2022 Dec;60(1):2098-2109. doi: 10.1080/13880209.2022.2132029. Pharm Biol. 2022. PMID: 36269032 Free PMC article.
-
The Effect of Eight Weeks of High-Intensity Interval Training on Follistatin Gene Expression in the Fast and Slow Twitch Muscles of Rats with Myocardial Infarction.Iran J Med Sci. 2024 Nov 1;49(11):716-723. doi: 10.30476/ijms.2024.99387.3141. eCollection 2024 Nov. Iran J Med Sci. 2024. PMID: 39678528 Free PMC article.
-
Angiotensin-(1-7) improves skeletal muscle regeneration.Eur J Transl Myol. 2023 Dec 15;33(4):12037. doi: 10.4081/ejtm.2023.12037. Eur J Transl Myol. 2023. PMID: 38112612 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
