

Refinement of 16S rRNA gene analysis for low biomass biospecimens
- PMID: 34031485
- PMCID: PMC8144411
- DOI: 10.1038/s41598-021-90226-2
Refinement of 16S rRNA gene analysis for low biomass biospecimens
Abstract
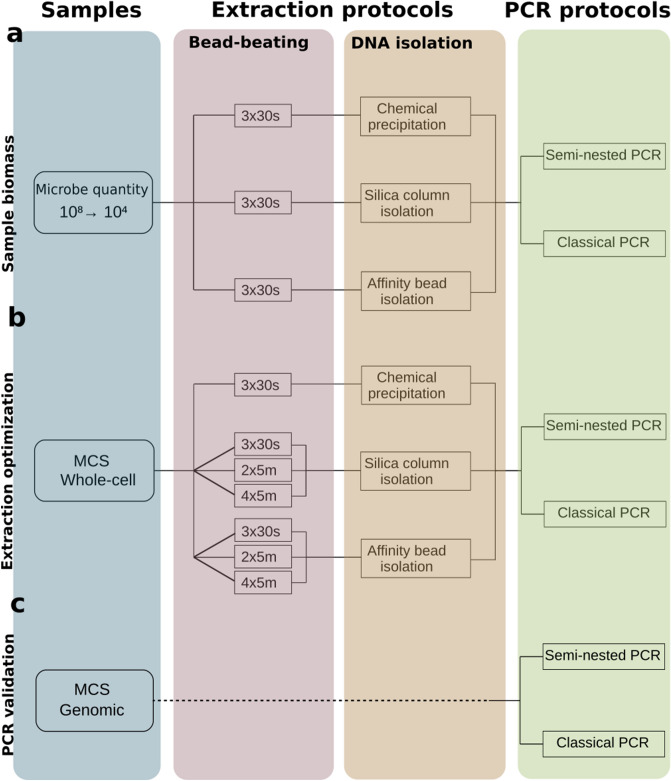
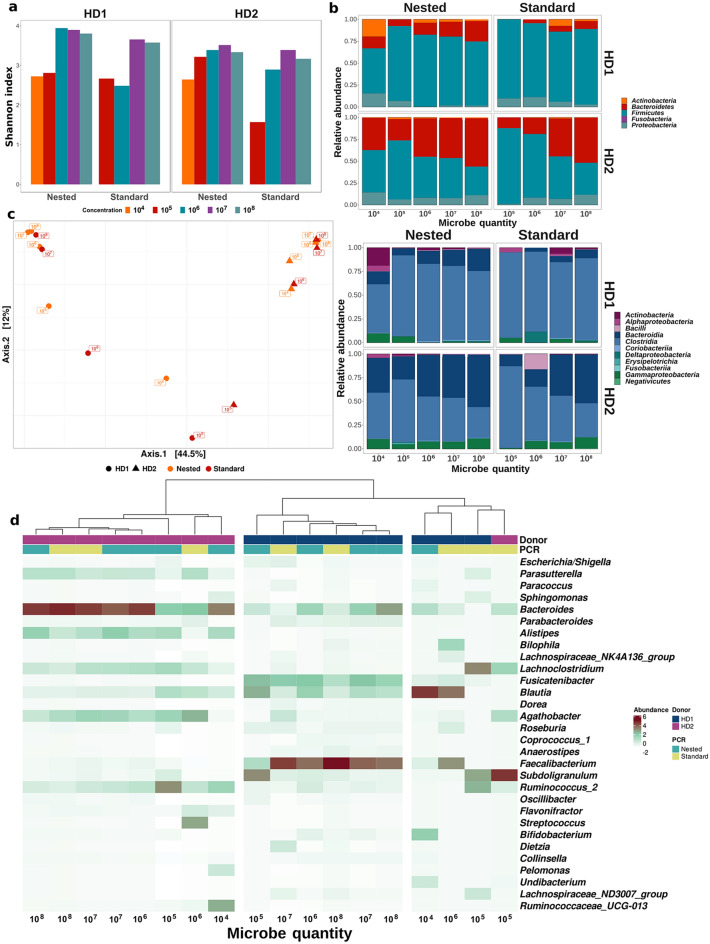
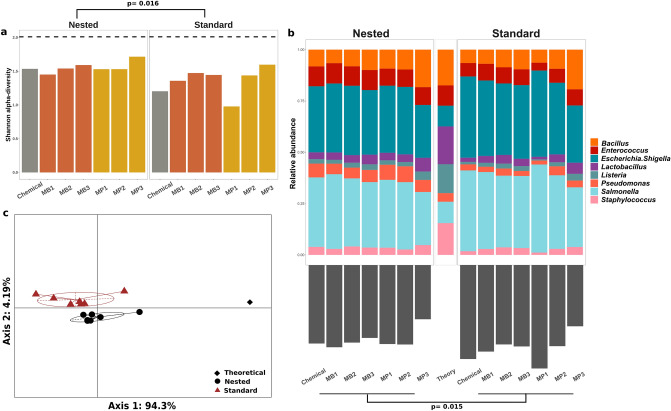
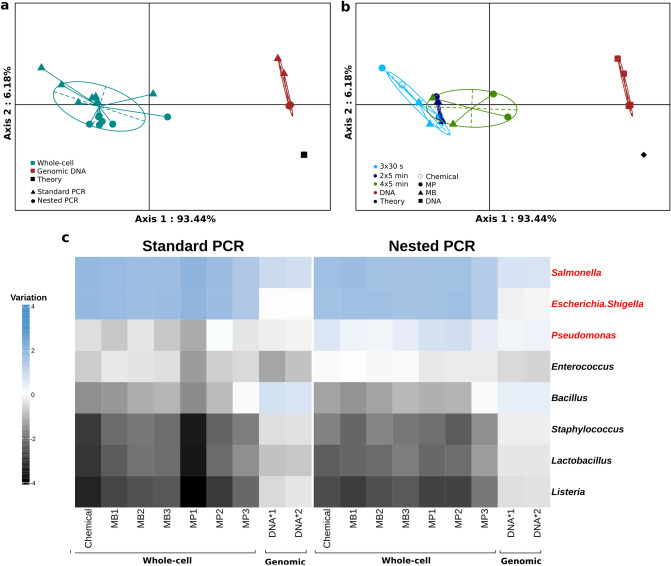
High-throughput phylogenetic 16S rRNA gene analysis has permitted to thoroughly delve into microbial community complexity and to understand host-microbiota interactions in health and disease. The analysis comprises sample collection and storage, genomic DNA extraction, 16S rRNA gene amplification, high-throughput amplicon sequencing and bioinformatic analysis. Low biomass microbiota samples (e.g. biopsies, tissue swabs and lavages) are receiving increasing attention, but optimal standardization for analysis of low biomass samples has yet to be developed. Here we tested the lower bacterial concentration required to perform 16S rRNA gene analysis using three different DNA extraction protocols, three different mechanical lysing series and two different PCR protocols. A mock microbiota community standard and low biomass samples (108, 107, 106, 105 and 104 microbes) from two healthy donor stools were employed to assess optimal sample processing for 16S rRNA gene analysis using paired-end Illumina MiSeq technology. Three DNA extraction protocols tested in our study performed similar with regards to representing microbiota composition, but extraction yield was better for silica columns compared to bead absorption and chemical precipitation. Furthermore, increasing mechanical lysing time and repetition did ameliorate the representation of bacterial composition. The most influential factor enabling appropriate representation of microbiota composition remains sample biomass. Indeed, bacterial densities below 106 cells resulted in loss of sample identity based on cluster analysis for all tested protocols. Finally, we excluded DNA extraction bias using a genomic DNA standard, which revealed that a semi-nested PCR protocol represented microbiota composition better than classical PCR. Based on our results, starting material concentration is an important limiting factor, highlighting the need to adapt protocols for dealing with low biomass samples. Our study suggests that the use of prolonged mechanical lysing, silica membrane DNA isolation and a semi-nested PCR protocol improve the analysis of low biomass samples. Using the improved protocol we report a lower limit of 106 bacteria per sample for robust and reproducible microbiota analysis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
