

Detecting adaptive introgression in human evolution using convolutional neural networks
- PMID: 34032215
- PMCID: PMC8192126
- DOI: 10.7554/eLife.64669
Detecting adaptive introgression in human evolution using convolutional neural networks
Abstract
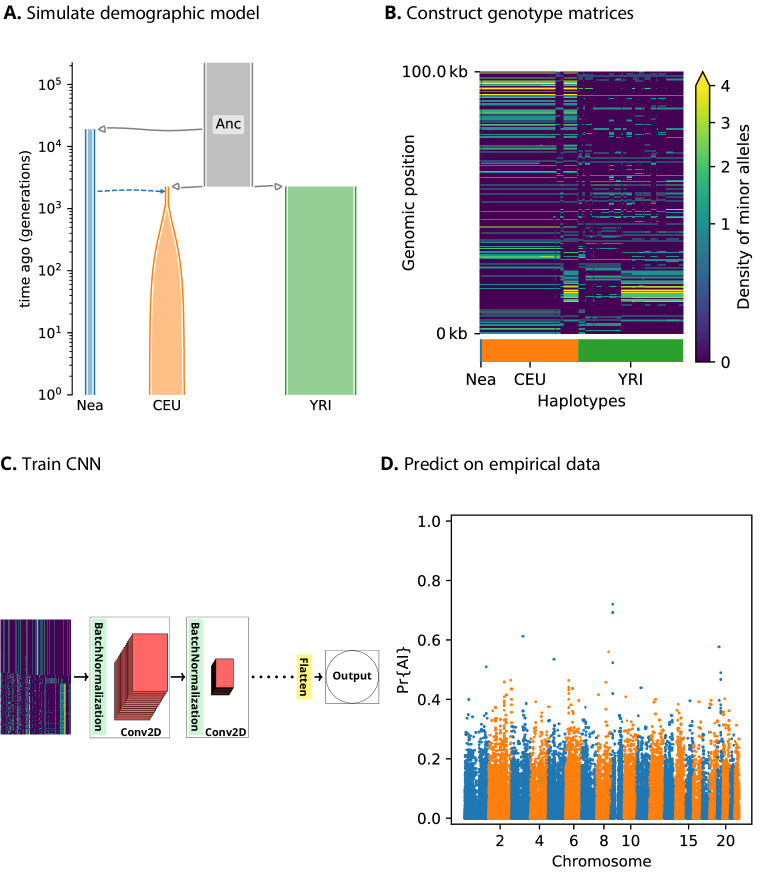
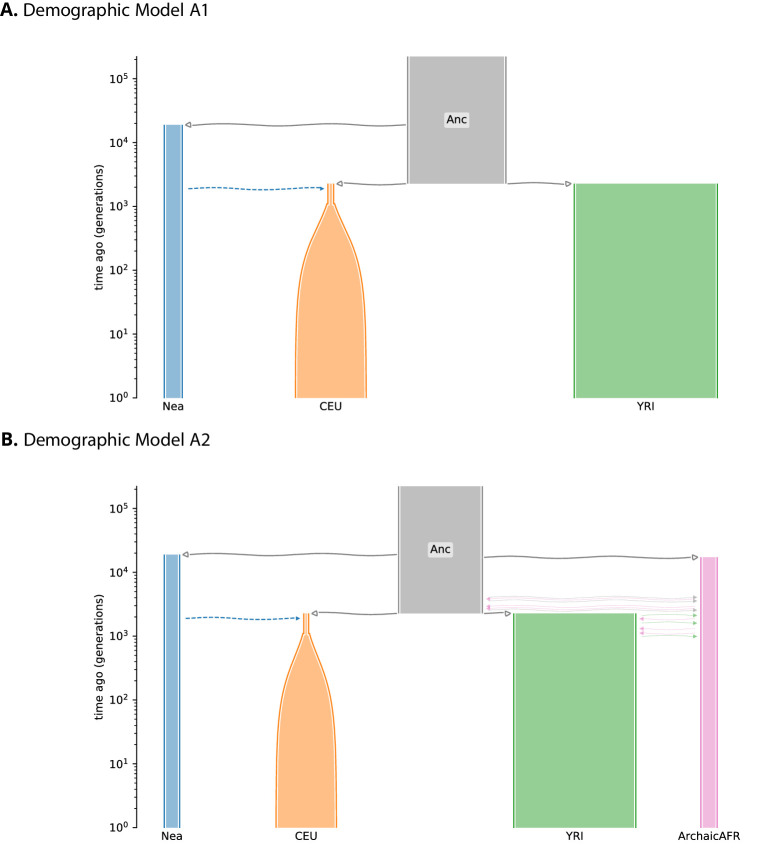
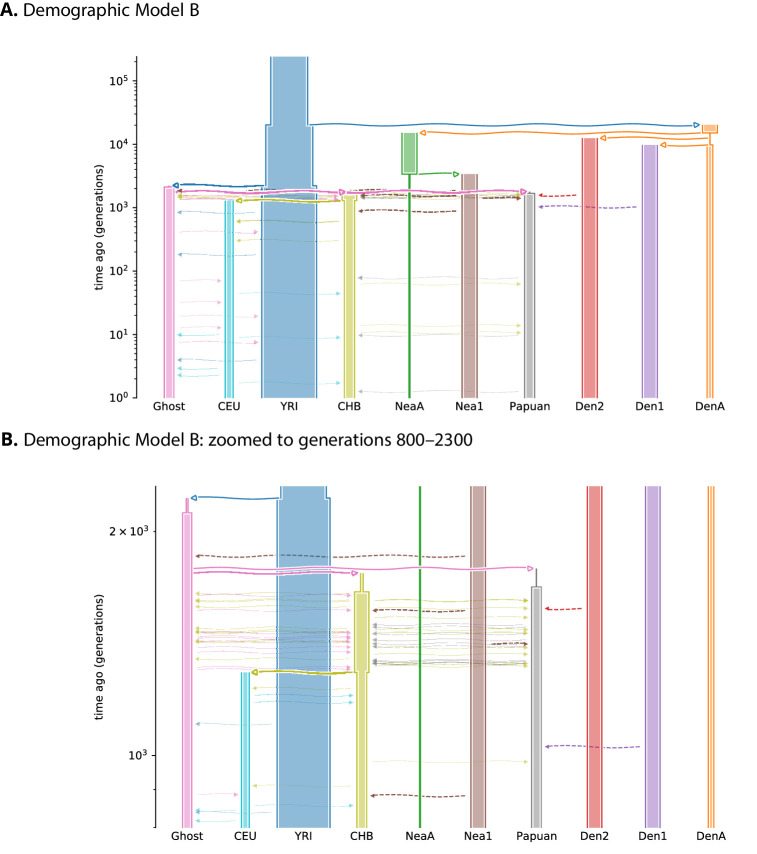
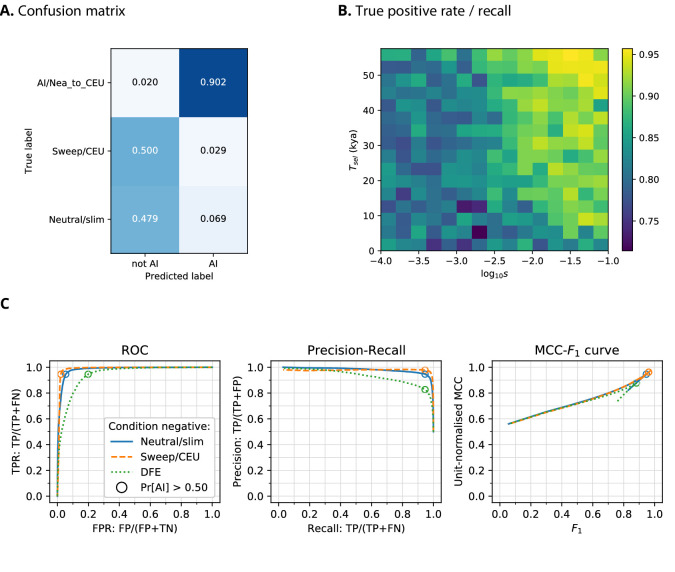
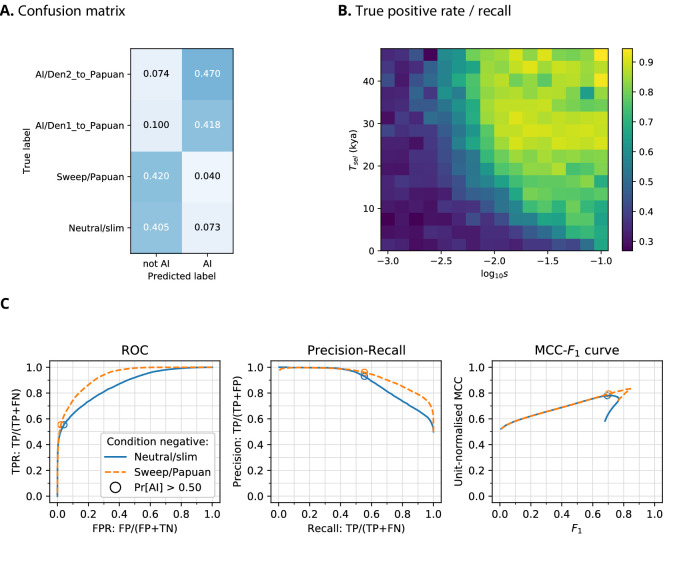
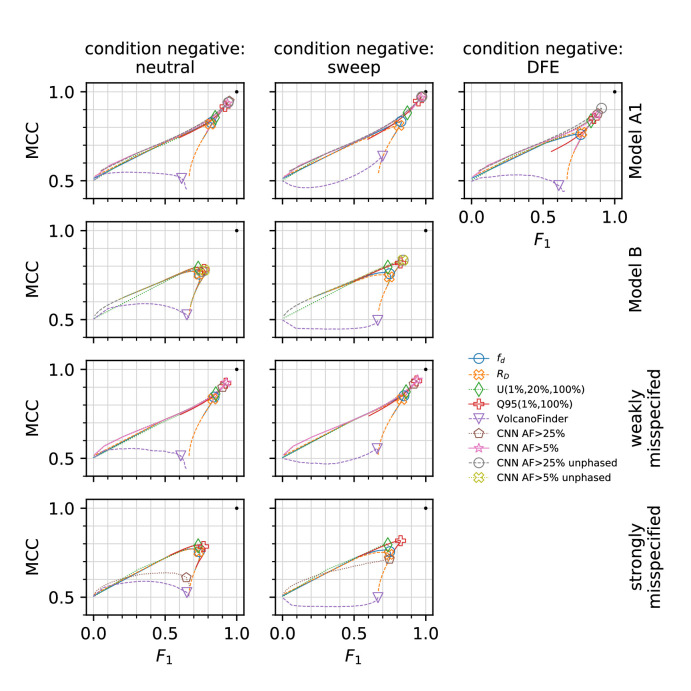
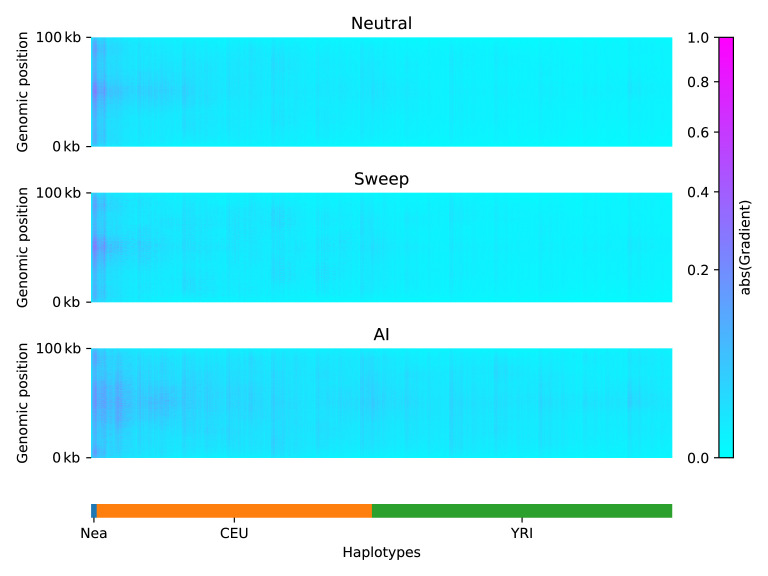
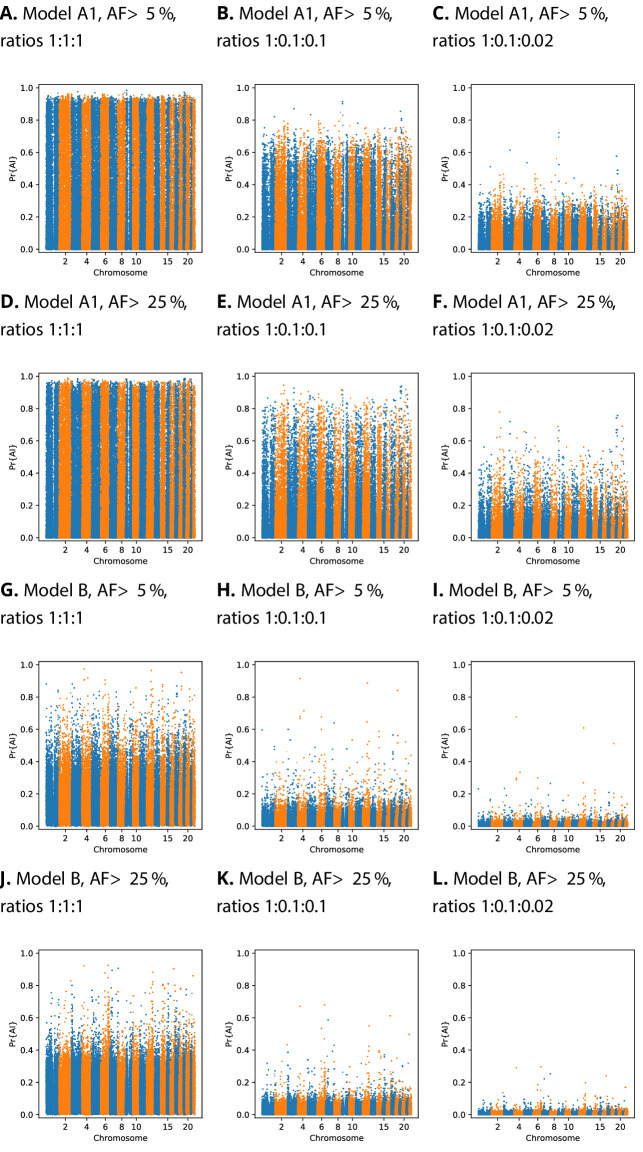
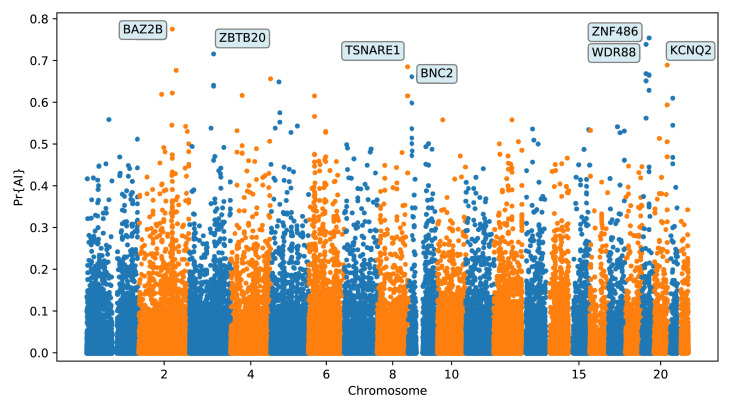
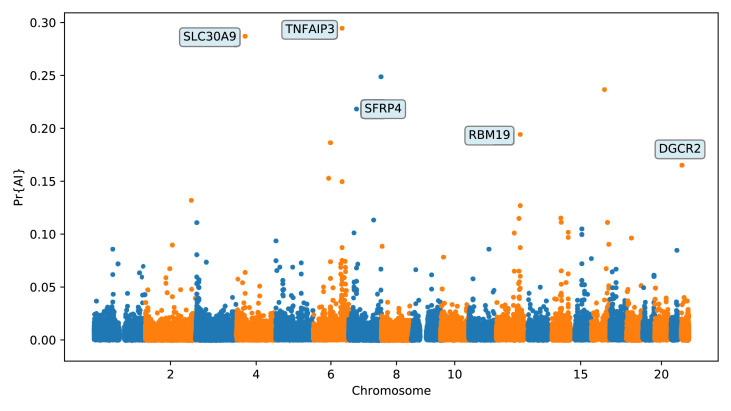
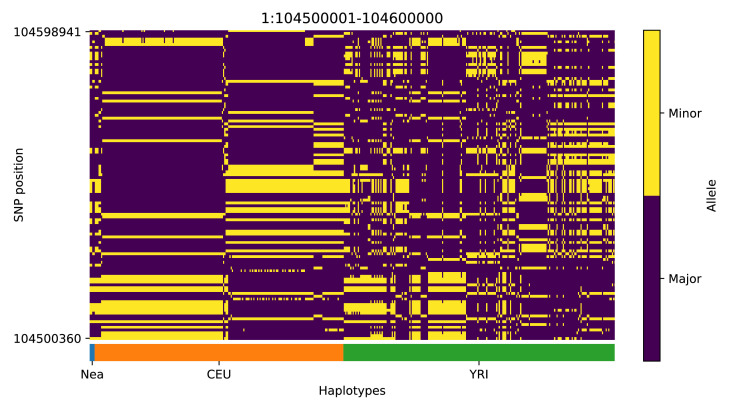
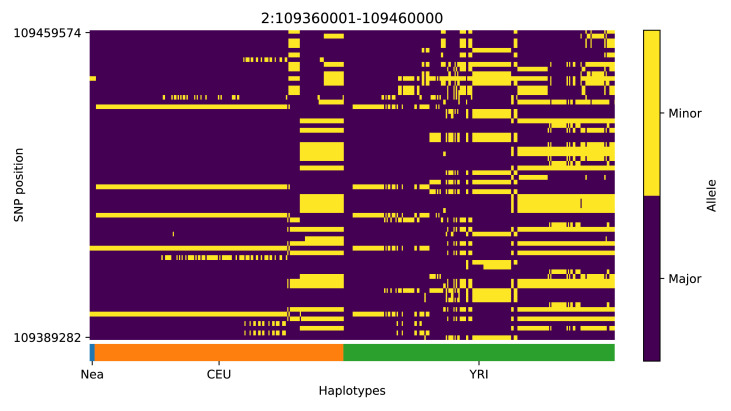
Studies in a variety of species have shown evidence for positively selected variants introduced into a population via introgression from another, distantly related population-a process known as adaptive introgression. However, there are few explicit frameworks for jointly modelling introgression and positive selection, in order to detect these variants using genomic sequence data. Here, we develop an approach based on convolutional neural networks (CNNs). CNNs do not require the specification of an analytical model of allele frequency dynamics and have outperformed alternative methods for classification and parameter estimation tasks in various areas of population genetics. Thus, they are potentially well suited to the identification of adaptive introgression. Using simulations, we trained CNNs on genotype matrices derived from genomes sampled from the donor population, the recipient population and a related non-introgressed population, in order to distinguish regions of the genome evolving under adaptive introgression from those evolving neutrally or experiencing selective sweeps. Our CNN architecture exhibits 95% accuracy on simulated data, even when the genomes are unphased, and accuracy decreases only moderately in the presence of heterosis. As a proof of concept, we applied our trained CNNs to human genomic datasets-both phased and unphased-to detect candidates for adaptive introgression that shaped our evolutionary history.
Keywords: adaptive introgression; computational biology; genetics; genomics; human; machine learning; simulation; systems biology.
© 2021, Gower et al.
Conflict of interest statement
GG, PP, MF, FR No competing interests declared
Figures
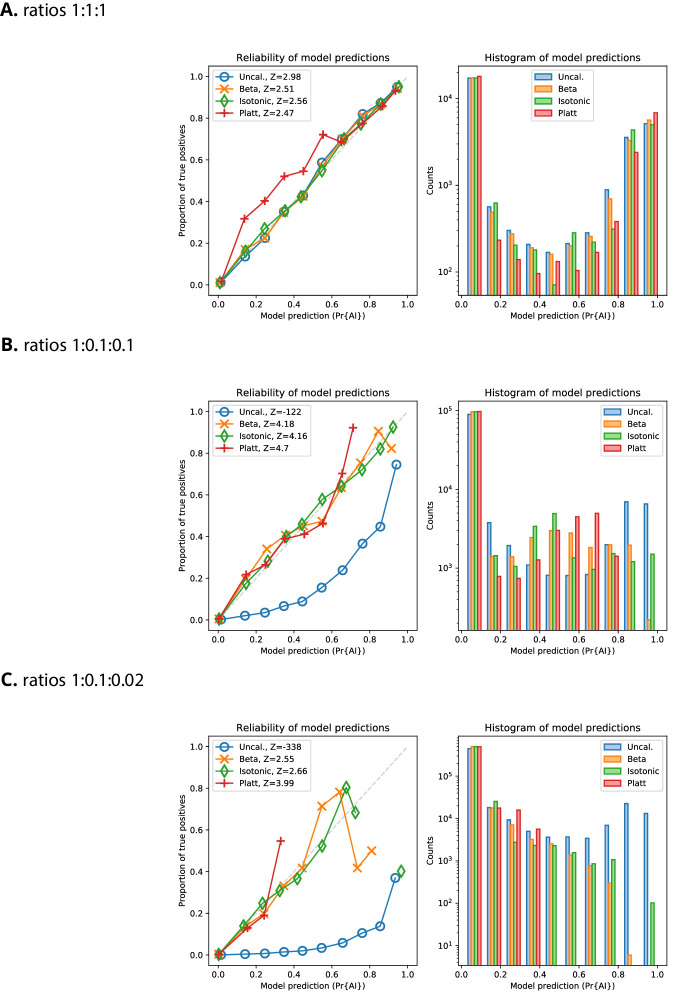
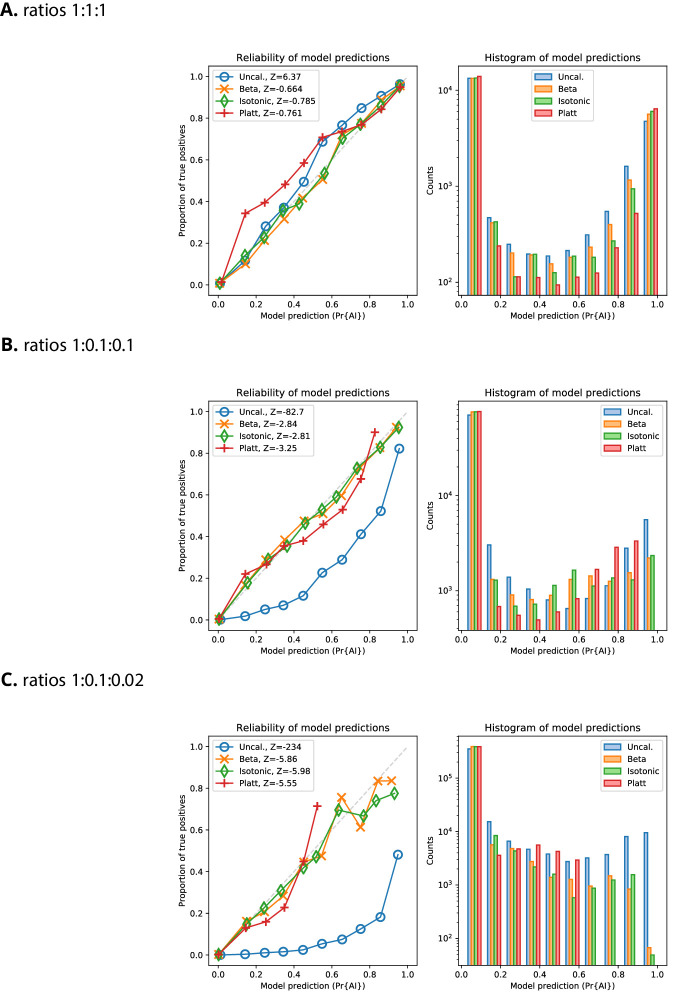
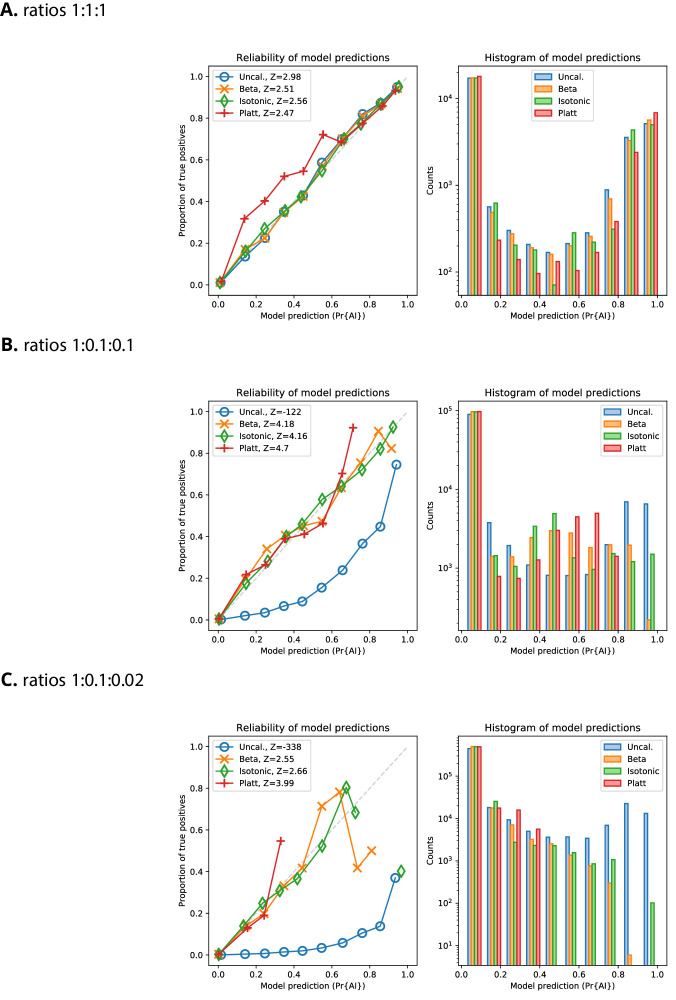
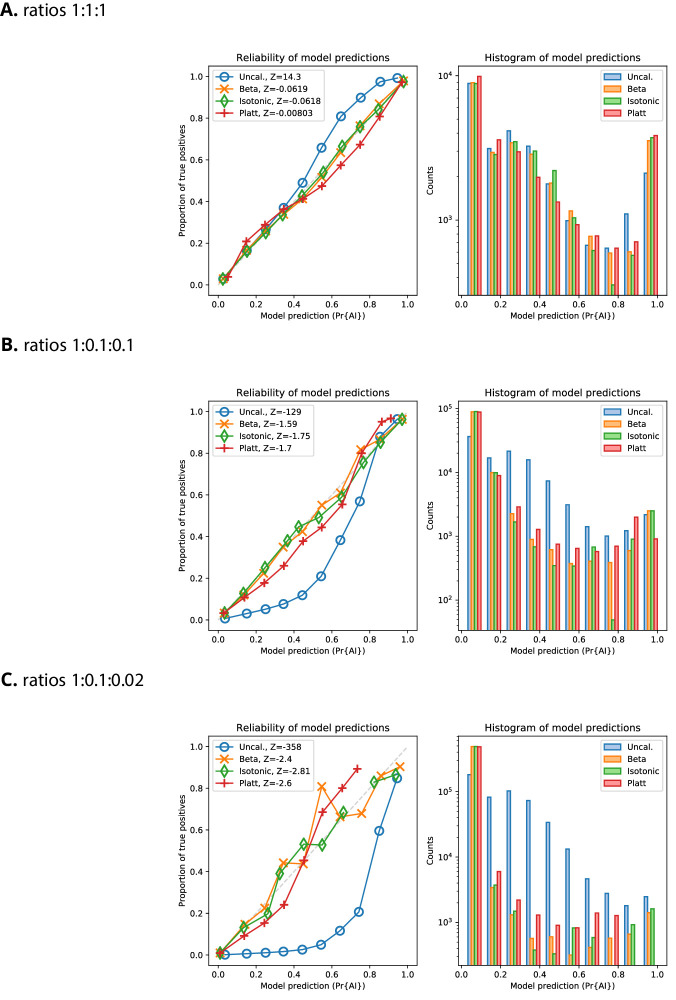
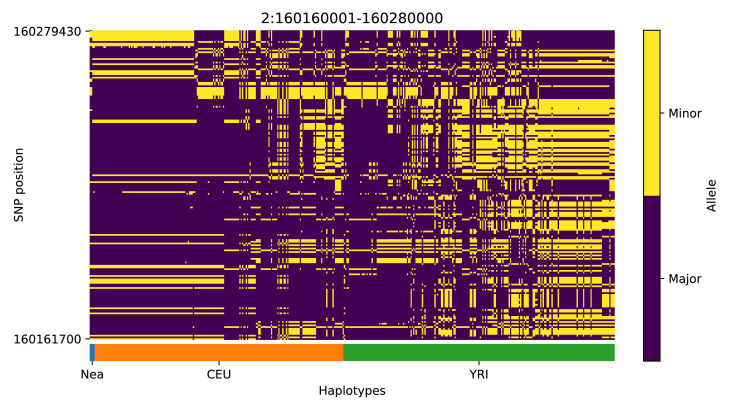
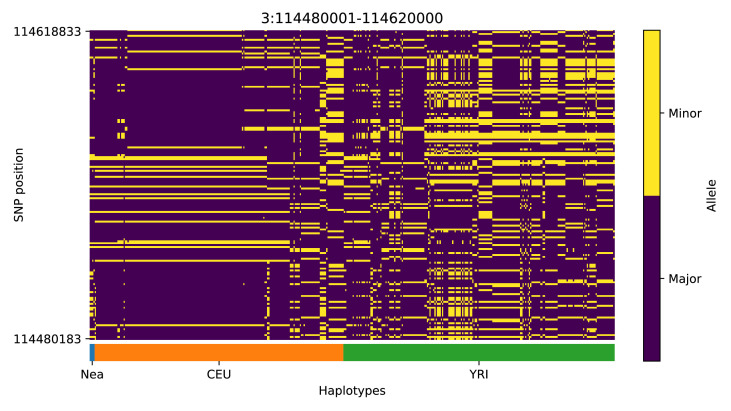



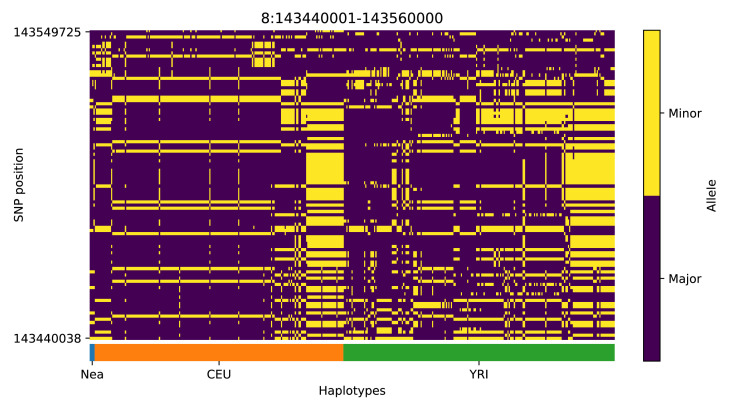
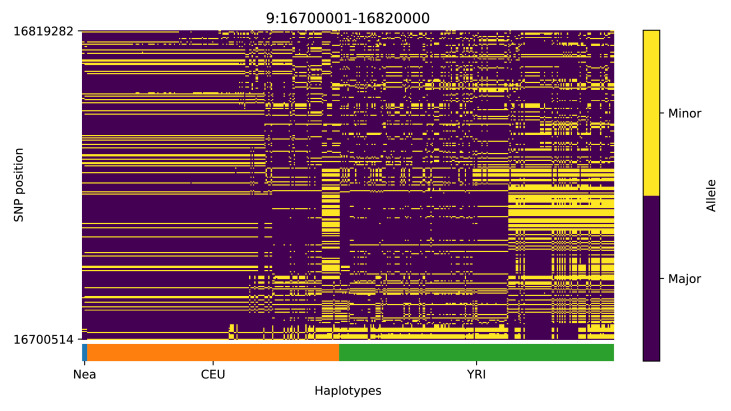





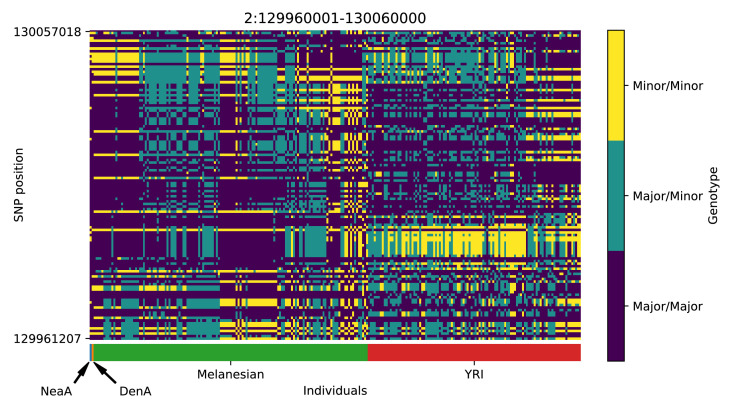
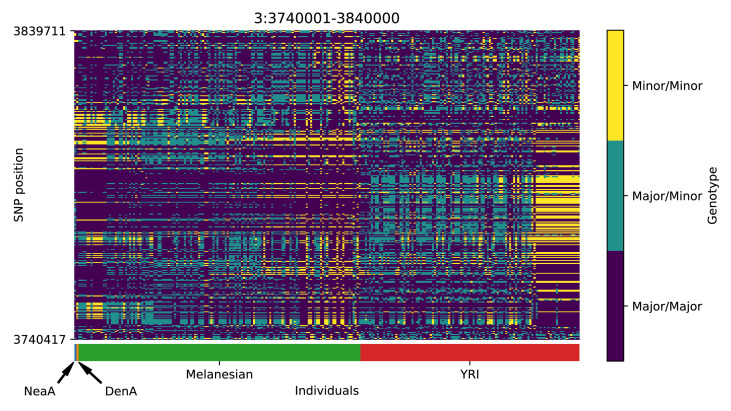
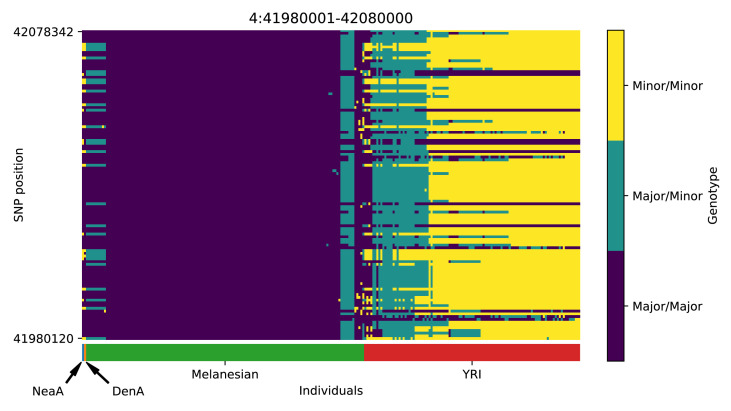
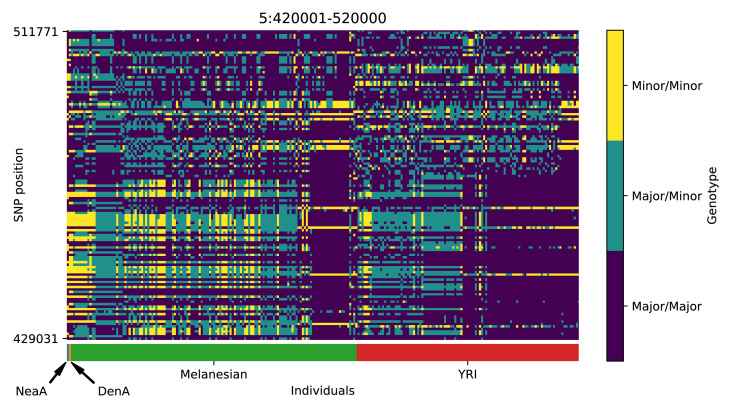
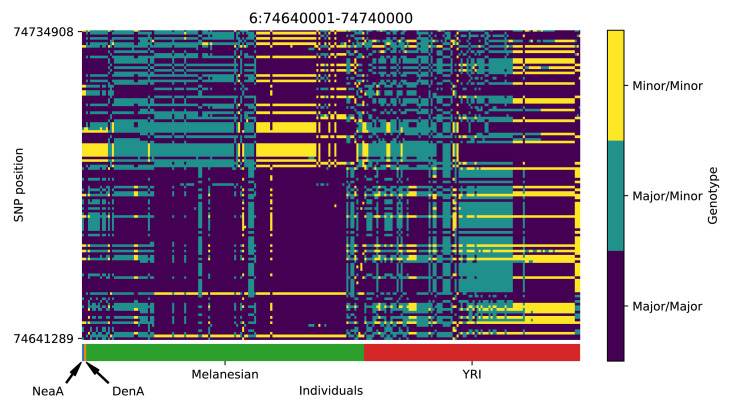
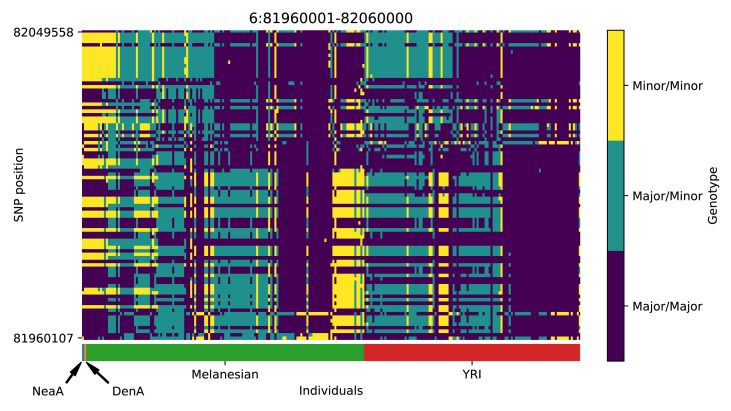

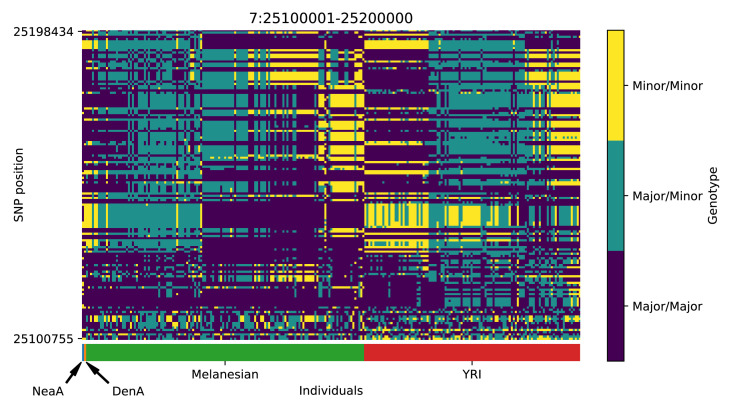
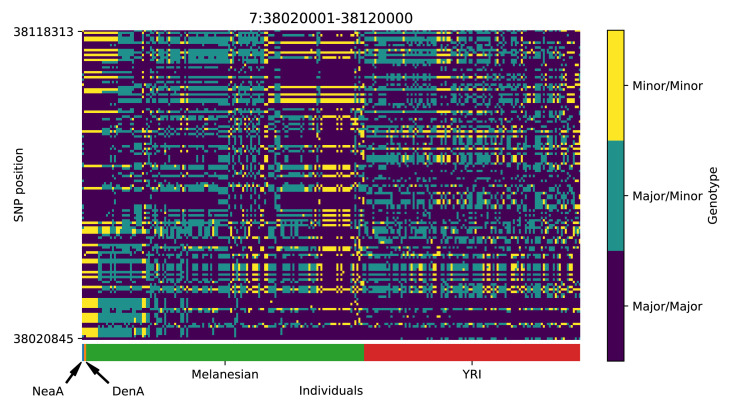
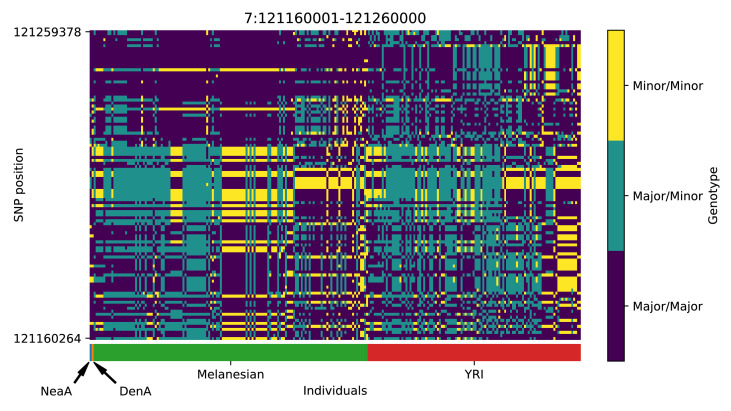

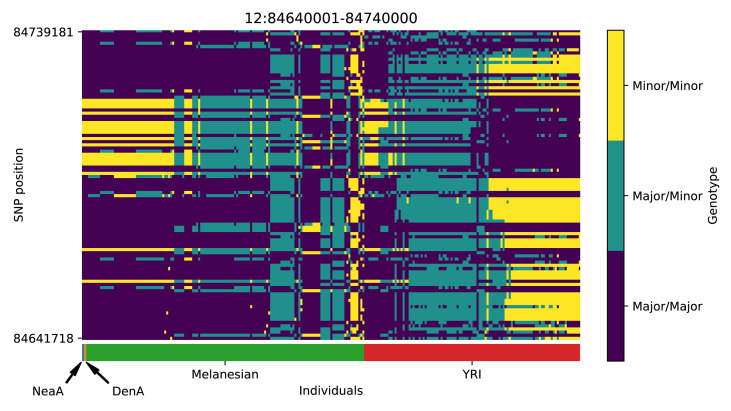

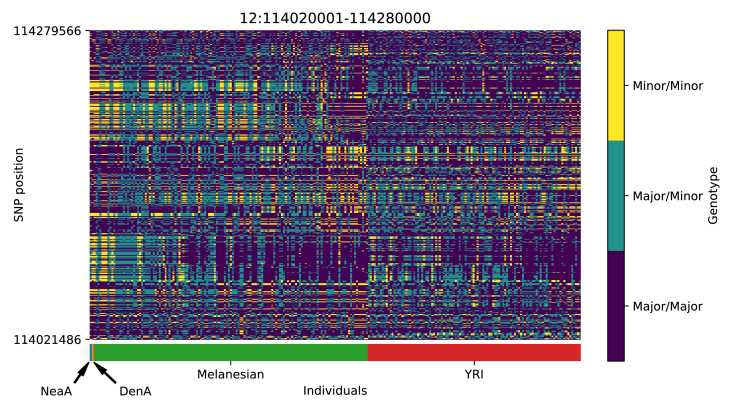
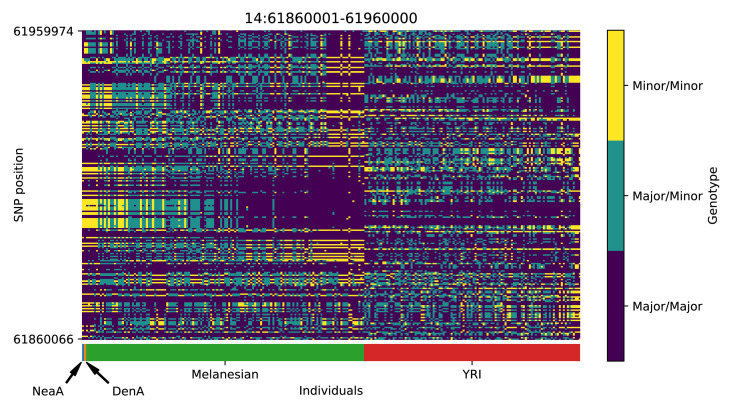

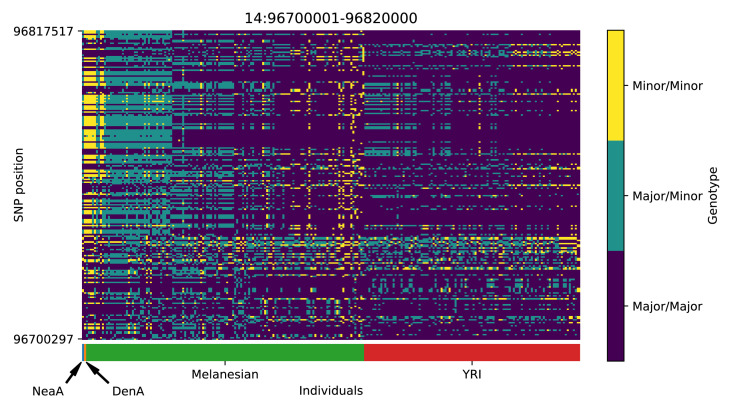
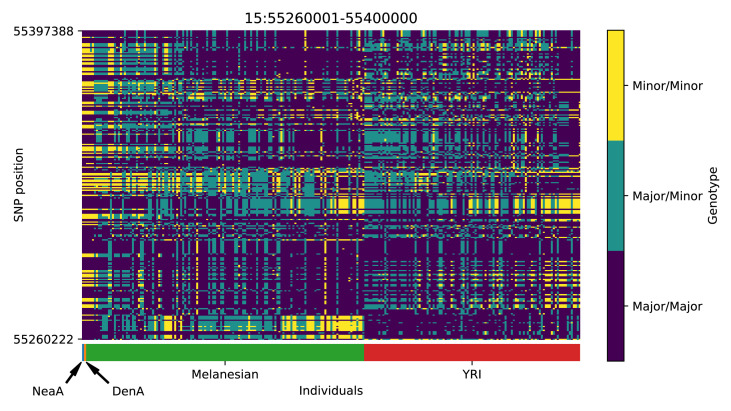
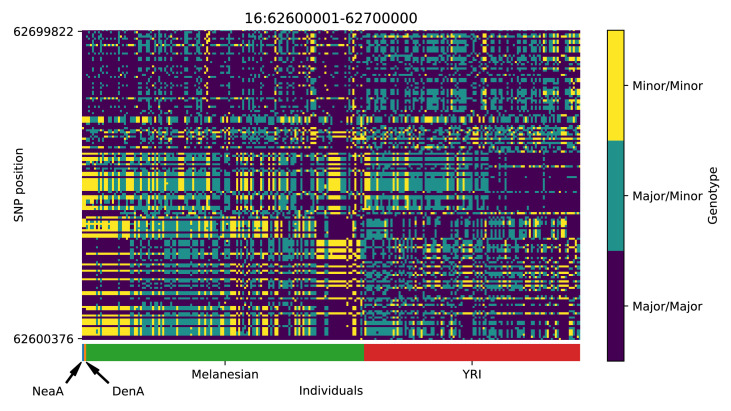
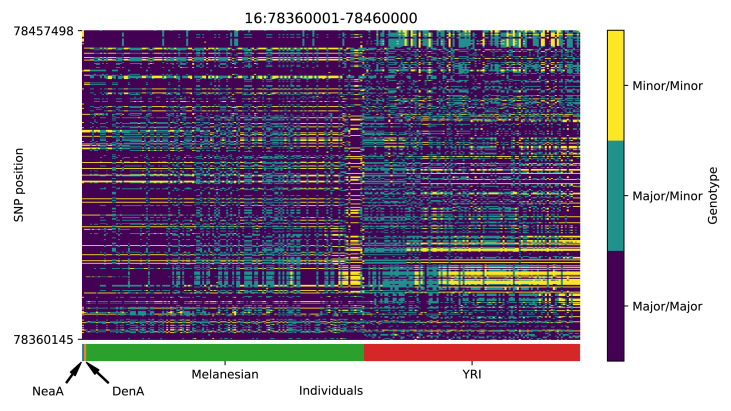
References
-
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, Ghemawat S, Goodfellow I, Harp A, Irving G, Isard M, Jia Y, Jozefowicz R, Kaiser L, Kudlur M, Levenberg J, Mané D, Monga R, Moore S, Murray D, Olah C, Schuster M, Shlens J, Steiner B, Sutskever I, Talwar K, Tucker P, Vanhoucke V, Vasudevan V, Viégas F, Vinyals O, Warden P, Wattenberg M, Wicke M, Yu Y, Zheng X. TensorFlow: large-scale machine learning on heterogeneous systems. arXiv. 2015 https://arxiv.org/abs/1603.04467
-
- Adrion JR, Cole CB, Dukler N, Galloway JG, Gladstein AL, Gower G, Kyriazis CC, Ragsdale AP, Tsambos G, Baumdicker F, Carlson J, Cartwright RA, Durvasula A, Gronau I, Kim BY, McKenzie P, Messer PW, Noskova E, Ortega-Del Vecchyo D, Racimo F, Struck TJ, Gravel S, Gutenkunst RN, Lohmueller KE, Ralph PL, Schrider DR, Siepel A, Kelleher J, Kern AD. A community-maintained standard library of population genetic models. eLife. 2020a;9:e54967. doi: 10.7554/eLife.54967. - DOI - PMC - PubMed
-
- Aggarwal CC. Neural Networks and Deep Learning. Springer; 2018. - DOI
-
- Alaa AM, van der Schaar M. Demystifying black-box models with symbolic metamodels. In: Wallach H, Larochelle H, Beygelzimer A, Alché-Buc F. d, Fox E, Garnett R, editors. Advances in Neural Information Processing Systems 32. Curran Associates, Inc; 2019. pp. 11304–11314.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
