

Genome-wide DNA mutations in Arabidopsis plants after multigenerational exposure to high temperatures
- PMID: 34034794
- PMCID: PMC8145854
- DOI: 10.1186/s13059-021-02381-4
Genome-wide DNA mutations in Arabidopsis plants after multigenerational exposure to high temperatures
Abstract
Background: Elevated temperatures can cause physiological, biochemical, and molecular responses in plants that can greatly affect their growth and development. Mutations are the most fundamental force driving biological evolution. However, how long-term elevations in temperature influence the accumulation of mutations in plants remains unknown.
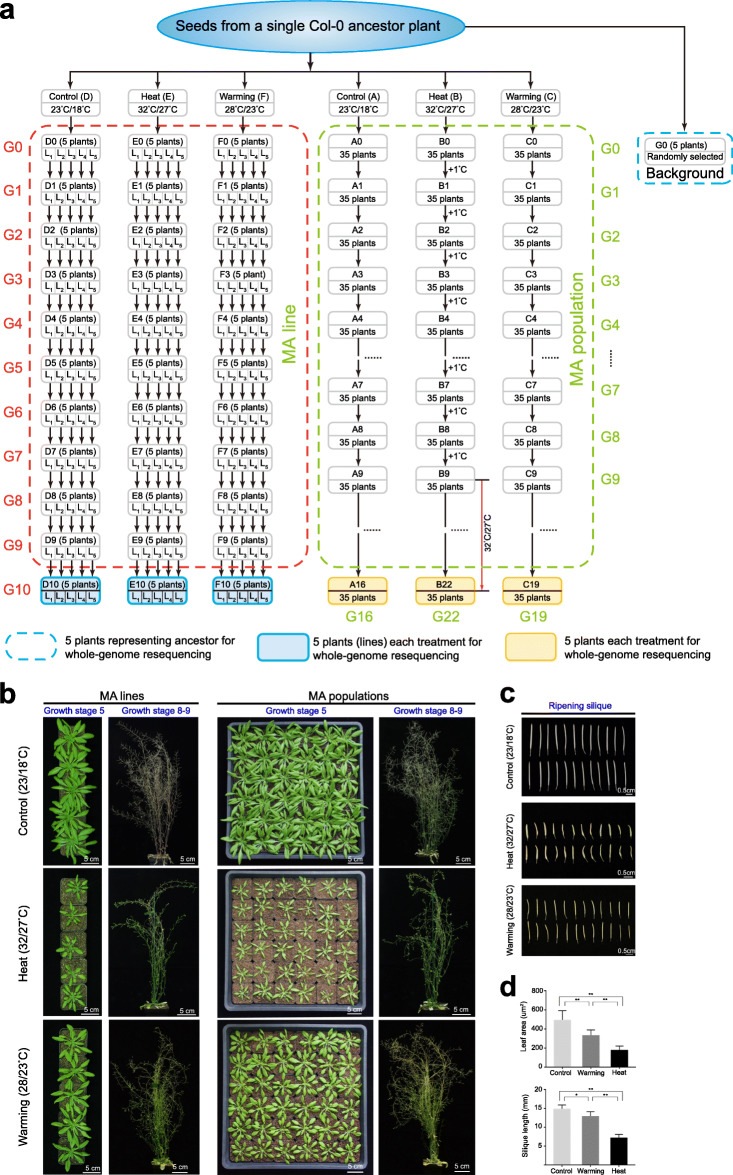
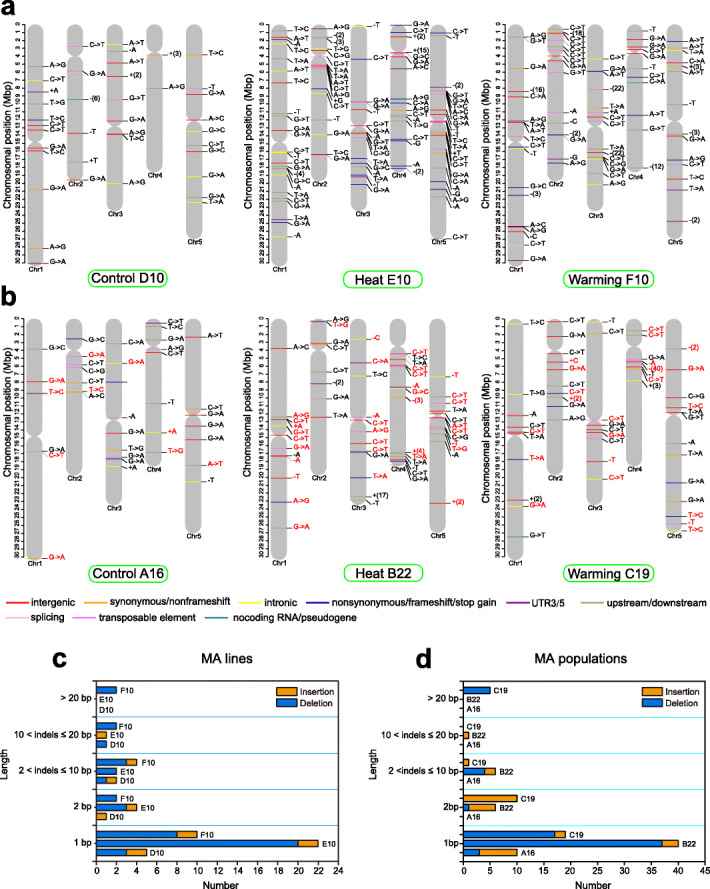
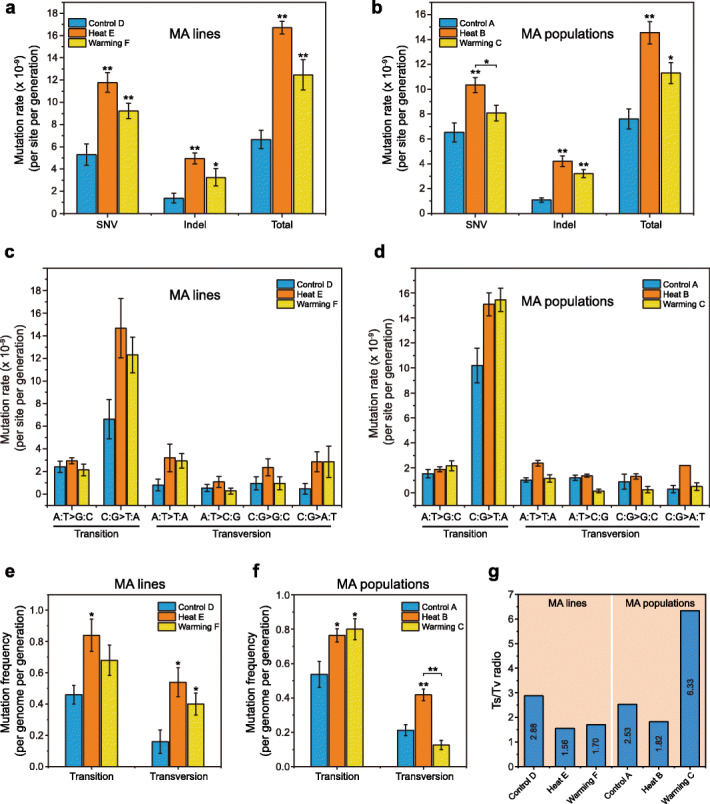
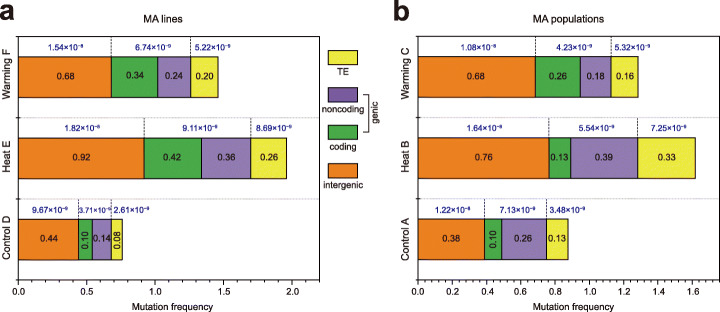
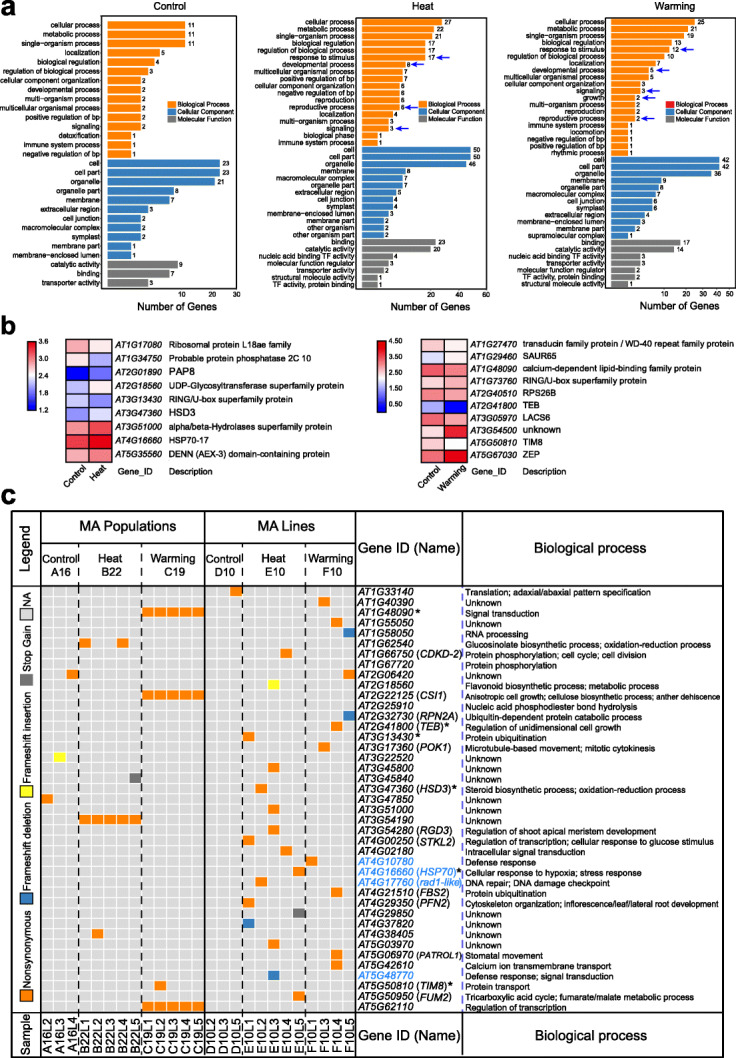
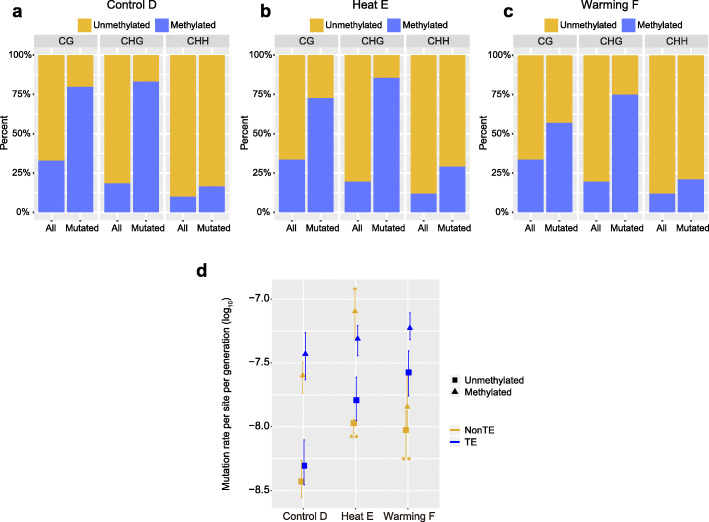
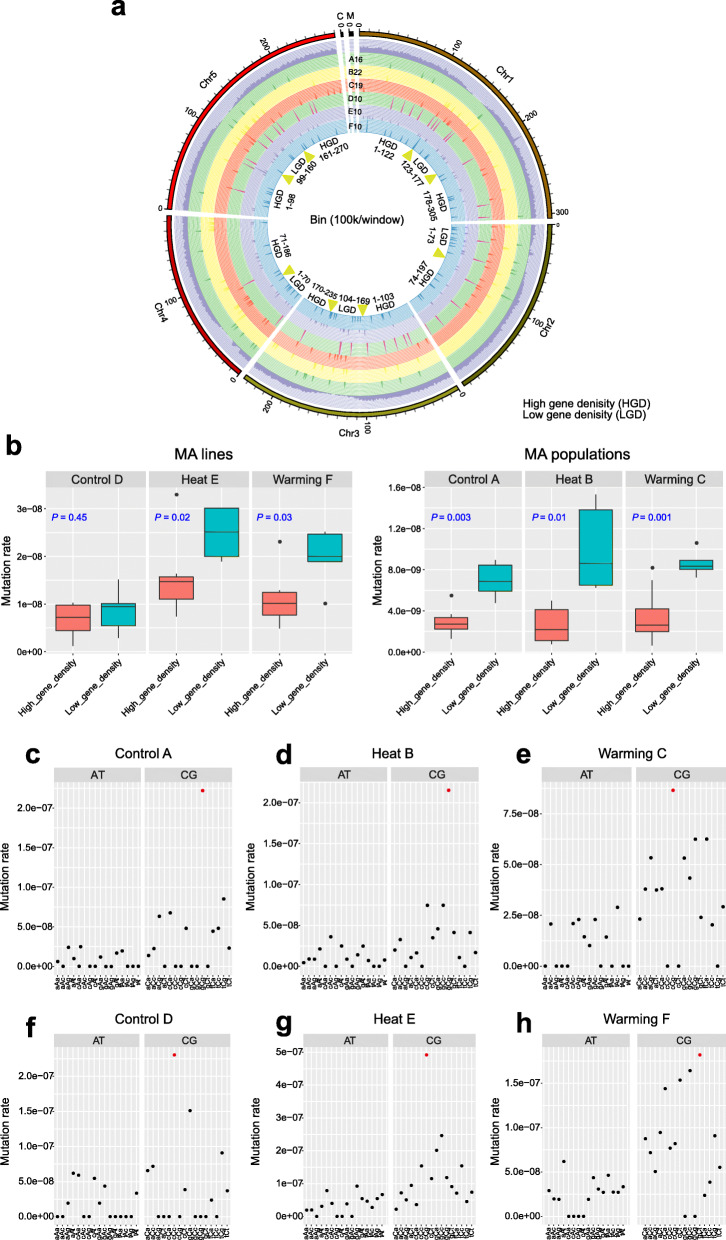
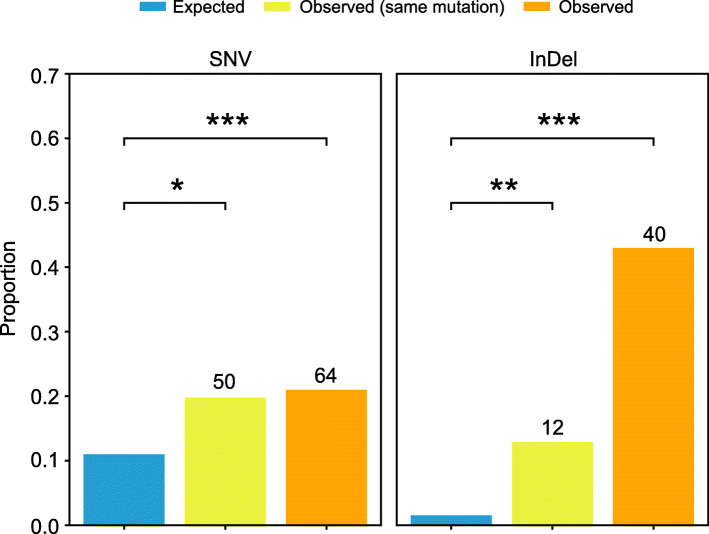
Results: Multigenerational exposure of Arabidopsis MA (mutation accumulation) lines and MA populations to extreme heat and moderate warming results in significantly increased mutation rates in single-nucleotide variants (SNVs) and small indels. We observe distinctive mutational spectra under extreme and moderately elevated temperatures, with significant increases in transition and transversion frequencies. Mutation occurs more frequently in intergenic regions, coding regions, and transposable elements in plants grown under elevated temperatures. At elevated temperatures, more mutations accumulate in genes associated with defense responses, DNA repair, and signaling. Notably, the distribution patterns of mutations among all progeny differ between MA populations and MA lines, suggesting that stronger selection effects occurred in populations. Methylation is observed more frequently at mutation sites, indicating its contribution to the mutation process at elevated temperatures. Mutations occurring within the same genome under elevated temperatures are significantly biased toward low gene density regions, special trinucleotides, tandem repeats, and adjacent simple repeats. Additionally, mutations found in all progeny overlap significantly with genetic variations reported in 1001 Genomes, suggesting non-uniform distribution of de novo mutations through the genome.
Conclusion: Collectively, our results suggest that elevated temperatures can accelerate the accumulation, and alter the molecular profiles, of DNA mutations in plants, thus providing significant insight into how environmental temperatures fuel plant evolution.
Keywords: Arabidopsis thaliana; Heat; Molecular evolution; Mutation accumulation; Mutation bias; Mutation rate; Mutation spectrum; Transposable element; Warming.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations.Genome Res. 2014 Nov;24(11):1821-9. doi: 10.1101/gr.177659.114. Epub 2014 Oct 14. Genome Res. 2014. PMID: 25314969 Free PMC article.
-
Transcription-related mutations and GC content drive variation in nucleotide substitution rates across the genomes of Arabidopsis thaliana and Arabidopsis lyrata.BMC Evol Biol. 2007 Apr 23;7:66. doi: 10.1186/1471-2148-7-66. BMC Evol Biol. 2007. PMID: 17451608 Free PMC article.
-
Fine-Grained Analysis of Spontaneous Mutation Spectrum and Frequency in Arabidopsis thaliana.Genetics. 2019 Feb;211(2):703-714. doi: 10.1534/genetics.118.301721. Epub 2018 Dec 4. Genetics. 2019. PMID: 30514707 Free PMC article.
-
Control of development and transposon movement by DNA methylation in Arabidopsis thaliana.Cold Spring Harb Symp Quant Biol. 2004;69:139-43. doi: 10.1101/sqb.2004.69.139. Cold Spring Harb Symp Quant Biol. 2004. PMID: 16117643 Review. No abstract available.
-
Diversity and evolution of transposable elements in Arabidopsis.Chromosome Res. 2014 Jun;22(2):203-16. doi: 10.1007/s10577-014-9418-8. Chromosome Res. 2014. PMID: 24801342 Review.
Cited by
-
Machine Learning for Plant Stress Modeling: A Perspective towards Hormesis Management.Plants (Basel). 2022 Apr 2;11(7):970. doi: 10.3390/plants11070970. Plants (Basel). 2022. PMID: 35406950 Free PMC article. Review.
-
Reply to: Re-evaluating evidence for adaptive mutation rate variation.Nature. 2023 Jul;619(7971):E57-E60. doi: 10.1038/s41586-023-06315-x. Nature. 2023. PMID: 37495874 Free PMC article. No abstract available.
-
Genome Sequences of Two Strains of Prototheca wickerhamii Provide Insight Into the Protothecosis Evolution.Front Cell Infect Microbiol. 2022 Feb 2;12:797017. doi: 10.3389/fcimb.2022.797017. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35186789 Free PMC article.
-
Experimentally heat-induced transposition increases drought tolerance in Arabidopsis thaliana.New Phytol. 2022 Oct;236(1):182-194. doi: 10.1111/nph.18322. Epub 2022 Jul 9. New Phytol. 2022. PMID: 35715973 Free PMC article.
-
Morphometrics and Phylogenomics of Coca (Erythroxylum spp.) Illuminate Its Reticulate Evolution, With Implications for Taxonomy.Mol Biol Evol. 2024 Jul 3;41(7):msae114. doi: 10.1093/molbev/msae114. Mol Biol Evol. 2024. PMID: 38982580 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
