

Blood DNA methylation and COVID-19 outcomes
- PMID: 34034806
- PMCID: PMC8148415
- DOI: 10.1186/s13148-021-01102-9
Blood DNA methylation and COVID-19 outcomes
Abstract
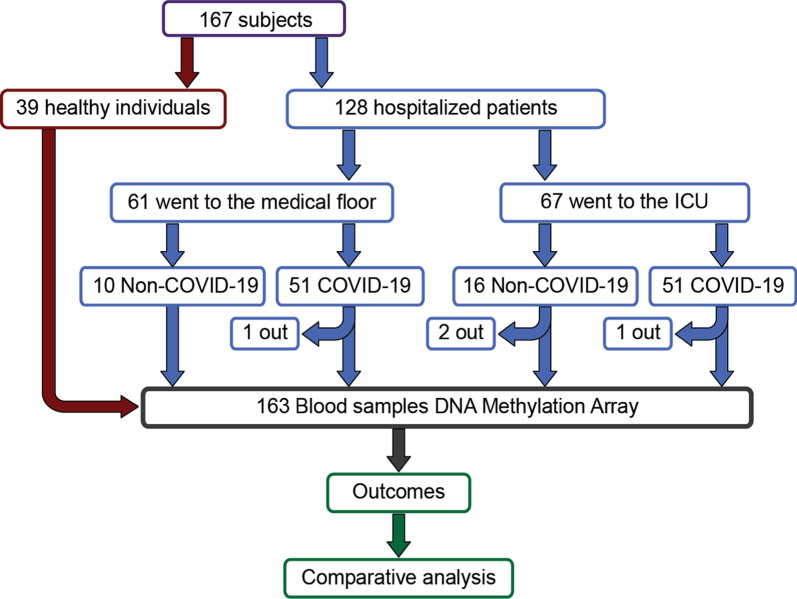
Background: There are no prior reports that compare differentially methylated regions of DNA in blood samples from COVID-19 patients to samples collected before the SARS-CoV-2 pandemic using a shared epigenotyping platform. We performed a genome-wide analysis of circulating blood DNA CpG methylation using the Infinium Human MethylationEPIC BeadChip on 124 blood samples from hospitalized COVID-19-positive and COVID-19-negative patients and compared these data with previously reported data from 39 healthy individuals collected before the pandemic. Prospective outcome measures such as COVID-19-GRAM risk-score and mortality were combined with methylation data.
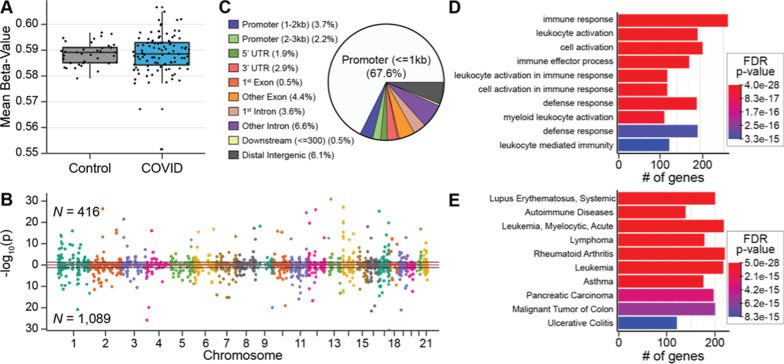
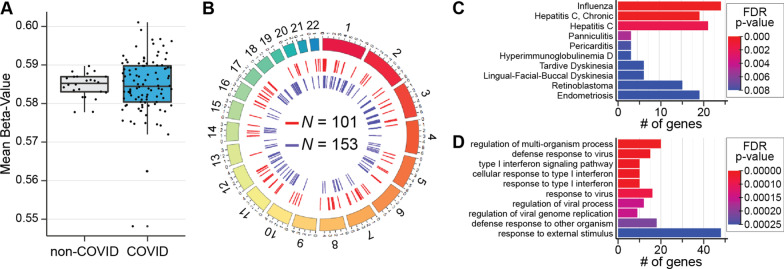
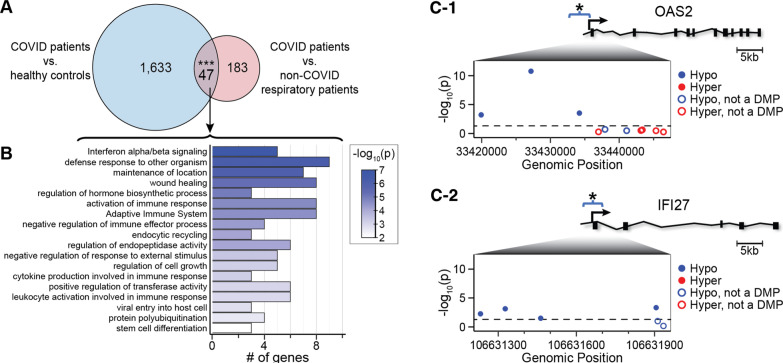
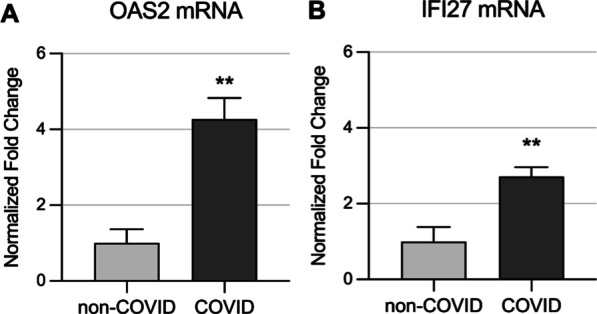
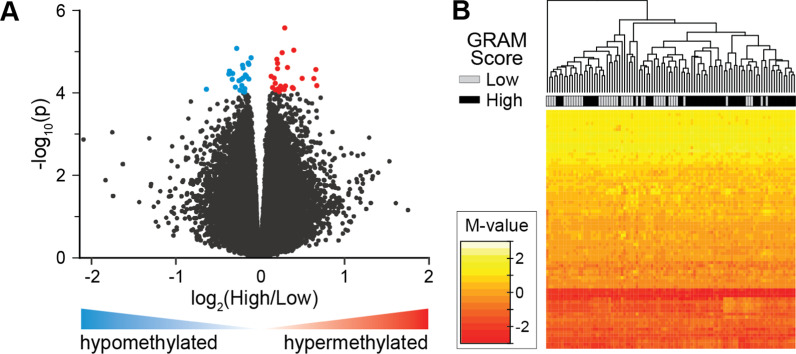
Results: Global mean methylation levels did not differ between COVID-19 patients and healthy pre-pandemic controls. About 75% of acute illness-associated differentially methylated regions were located near gene promoter regions and were hypo-methylated in comparison with healthy pre-pandemic controls. Gene ontology analyses revealed terms associated with the immune response to viral infections and leukocyte activation; and disease ontology analyses revealed a predominance of autoimmune disorders. Among COVID-19-positive patients, worse outcomes were associated with a prevailing hyper-methylated status. Recursive feature elimination identified 77 differentially methylated positions predictive of COVID-19 severity measured by the GRAM-risk score.
Conclusion: Our data contribute to the awareness that DNA methylation may influence the expression of genes that regulate COVID-19 progression and represent a targetable process in that setting.
Keywords: Acute respiratory distress syndrome (ARDS); COVID-19; DNA methylation; Gene expression; Mortality; Outcomes.
Conflict of interest statement
M.D.R. has financial relationships with CaroGen Corporation, and receives research funding from Gilead Sciences, outside of this work.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
