

Ectopic targeting of CG DNA methylation in Arabidopsis with the bacterial SssI methyltransferase
- PMID: 34035251
- PMCID: PMC8149686
- DOI: 10.1038/s41467-021-23346-y
Ectopic targeting of CG DNA methylation in Arabidopsis with the bacterial SssI methyltransferase
Abstract
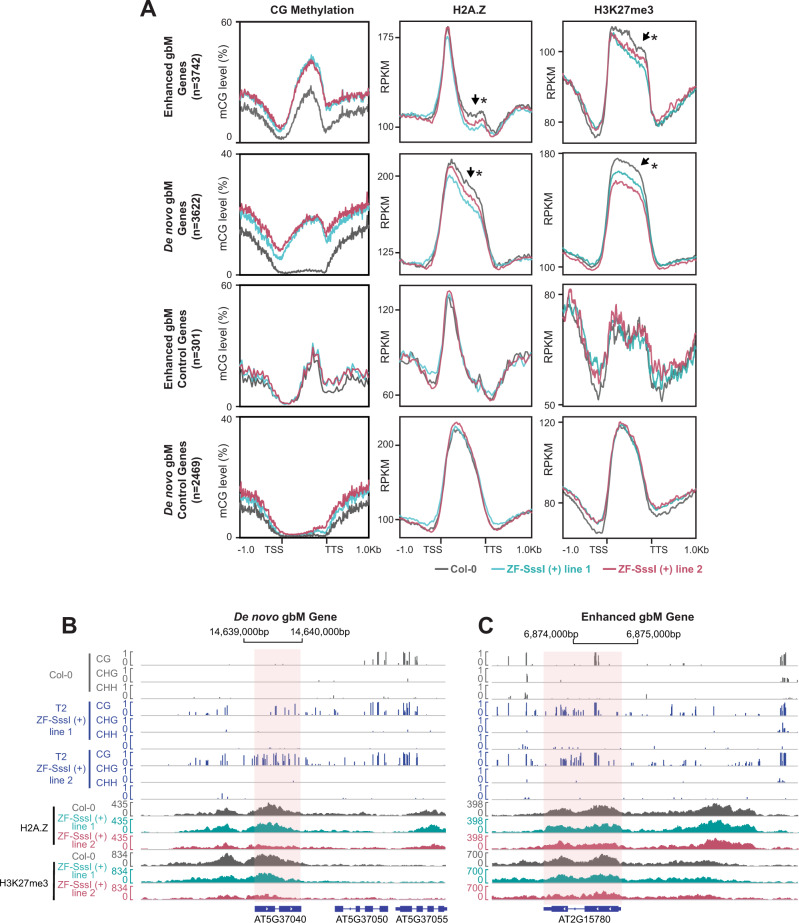
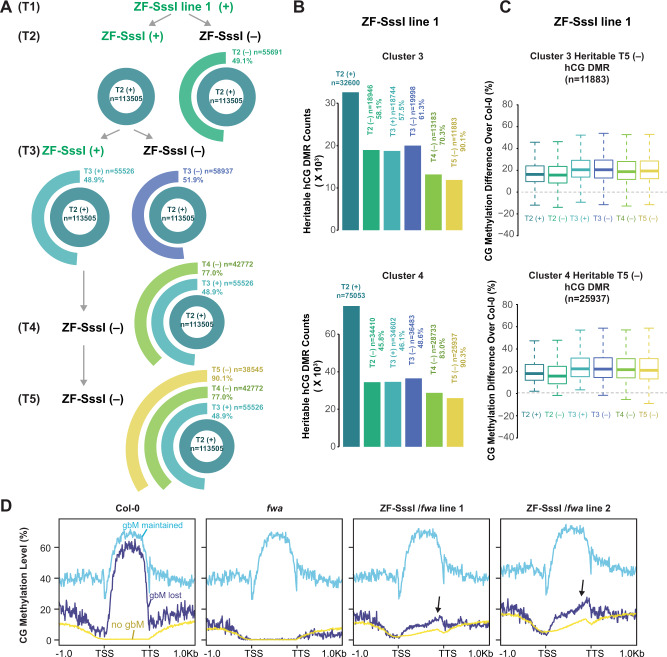
The ability to target epigenetic marks like DNA methylation to specific loci is important in both basic research and in crop plant engineering. However, heritability of targeted DNA methylation, how it impacts gene expression, and which epigenetic features are required for proper establishment are mostly unknown. Here, we show that targeting the CG-specific methyltransferase M.SssI with an artificial zinc finger protein can establish heritable CG methylation and silencing of a targeted locus in Arabidopsis. In addition, we observe highly heritable widespread ectopic CG methylation mainly over euchromatic regions. This hypermethylation shows little effect on transcription while it triggers a mild but significant reduction in the accumulation of H2A.Z and H3K27me3. Moreover, ectopic methylation occurs preferentially at less open chromatin that lacks positive histone marks. These results outline general principles of the heritability and interaction of CG methylation with other epigenomic features that should help guide future efforts to engineer epigenomes.
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase.Proc Natl Acad Sci U S A. 2021 Jun 8;118(23):e2125016118. doi: 10.1073/pnas.2125016118. Proc Natl Acad Sci U S A. 2021. PMID: 34074795 Free PMC article.
-
RNAi-independent de novo DNA methylation revealed in Arabidopsis mutants of chromatin remodeling gene DDM1.Plant J. 2012 Jun;70(5):750-8. doi: 10.1111/j.1365-313X.2012.04911.x. Epub 2012 Mar 6. Plant J. 2012. PMID: 22269081
-
The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA-directed DNA methylation in Arabidopsis thaliana.PLoS Genet. 2010 Oct 28;6(10):e1001182. doi: 10.1371/journal.pgen.1001182. PLoS Genet. 2010. PMID: 21060858 Free PMC article.
-
Silencing of transposons in plant genomes: kick them when they're down.Genome Biol. 2004;5(12):249. doi: 10.1186/gb-2004-5-12-249. Epub 2004 Nov 16. Genome Biol. 2004. PMID: 15575975 Free PMC article. Review.
-
siRNA-mediated chromatin maintenance and its function in Arabidopsis thaliana.Biochim Biophys Acta. 2011 Aug;1809(8):444-51. doi: 10.1016/j.bbagrm.2011.05.002. Epub 2011 May 13. Biochim Biophys Acta. 2011. PMID: 21605714 Review.
Cited by
-
RNA 5-Methylcytosine Modification Regulates Vegetative Development Associated with H3K27 Trimethylation in Arabidopsis.Adv Sci (Weinh). 2022 Nov 16;10(1):e2204885. doi: 10.1002/advs.202204885. Online ahead of print. Adv Sci (Weinh). 2022. PMID: 36382558 Free PMC article.
-
Prospects and challenges of epigenomics in crop improvement.Genes Genomics. 2022 Mar;44(3):251-257. doi: 10.1007/s13258-021-01187-9. Epub 2021 Nov 27. Genes Genomics. 2022. PMID: 34837632 Review.
-
DNA methylation underpins the epigenomic landscape regulating genome transcription in Arabidopsis.Genome Biol. 2022 Sep 20;23(1):197. doi: 10.1186/s13059-022-02768-x. Genome Biol. 2022. PMID: 36127735 Free PMC article.
-
The TRIPLE PHD FINGERS proteins are required for SWI/SNF complex-mediated +1 nucleosome positioning and transcription start site determination in Arabidopsis.Nucleic Acids Res. 2022 Oct 14;50(18):10399-10417. doi: 10.1093/nar/gkac826. Nucleic Acids Res. 2022. PMID: 36189880 Free PMC article.
-
Genome wide inherited modifications of the tomato epigenome by trans-activated bacterial CG methyltransferase.Cell Mol Life Sci. 2024 May 20;81(1):222. doi: 10.1007/s00018-024-05255-7. Cell Mol Life Sci. 2024. PMID: 38767725 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
