

Characterization of the public transit air microbiome and resistome reveals geographical specificity
- PMID: 34039416
- PMCID: PMC8157753
- DOI: 10.1186/s40168-021-01044-7
Characterization of the public transit air microbiome and resistome reveals geographical specificity
Abstract
Background: The public transit is a built environment with high occupant density across the globe, and identifying factors shaping public transit air microbiomes will help design strategies to minimize the transmission of pathogens. However, the majority of microbiome works dedicated to the public transit air are limited to amplicon sequencing, and our knowledge regarding the functional potentials and the repertoire of resistance genes (i.e. resistome) is limited. Furthermore, current air microbiome investigations on public transit systems are focused on single cities, and a multi-city assessment of the public transit air microbiome will allow a greater understanding of whether and how broad environmental, building, and anthropogenic factors shape the public transit air microbiome in an international scale. Therefore, in this study, the public transit air microbiomes and resistomes of six cities across three continents (Denver, Hong Kong, London, New York City, Oslo, Stockholm) were characterized.
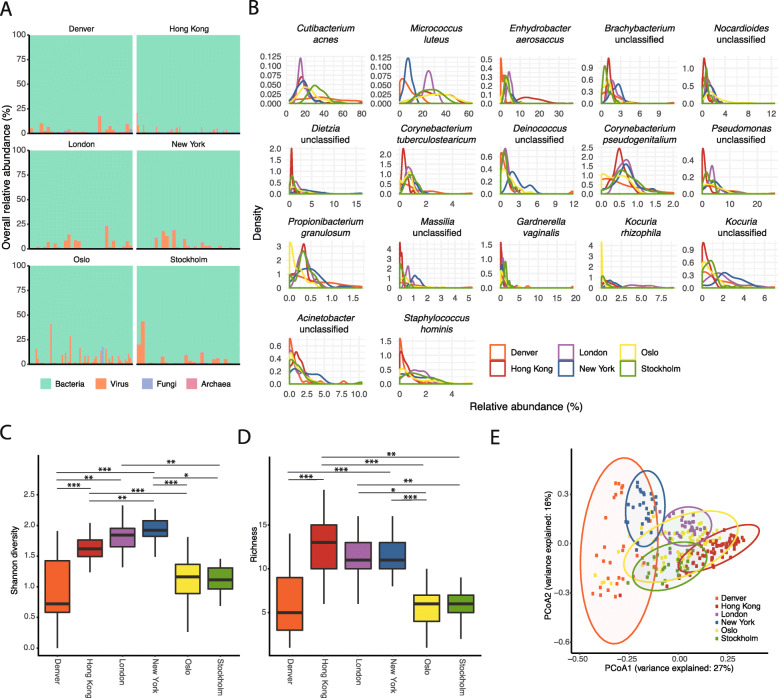
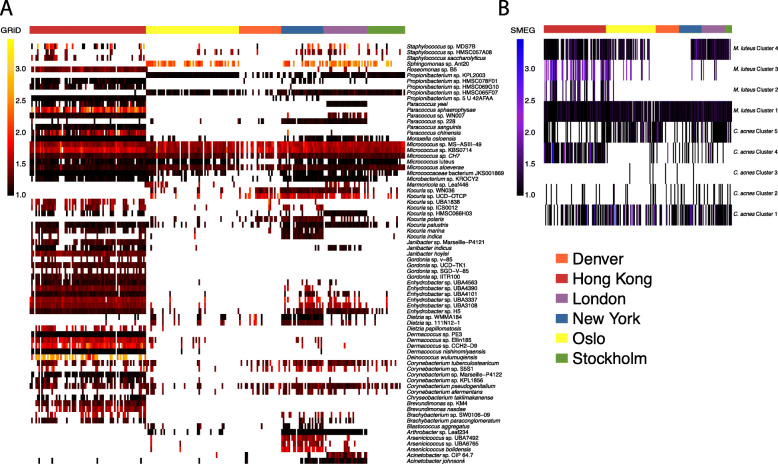
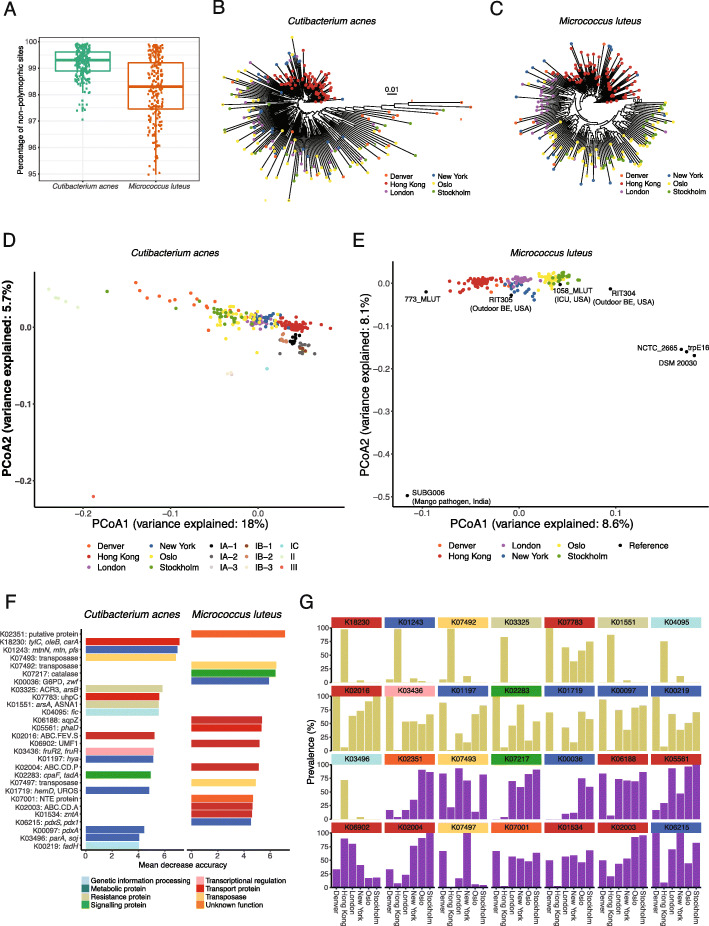
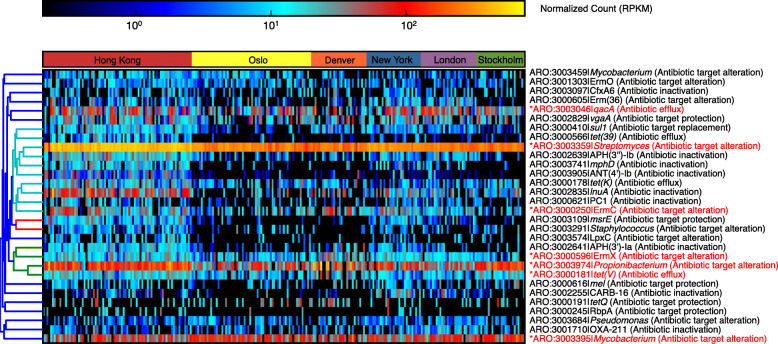
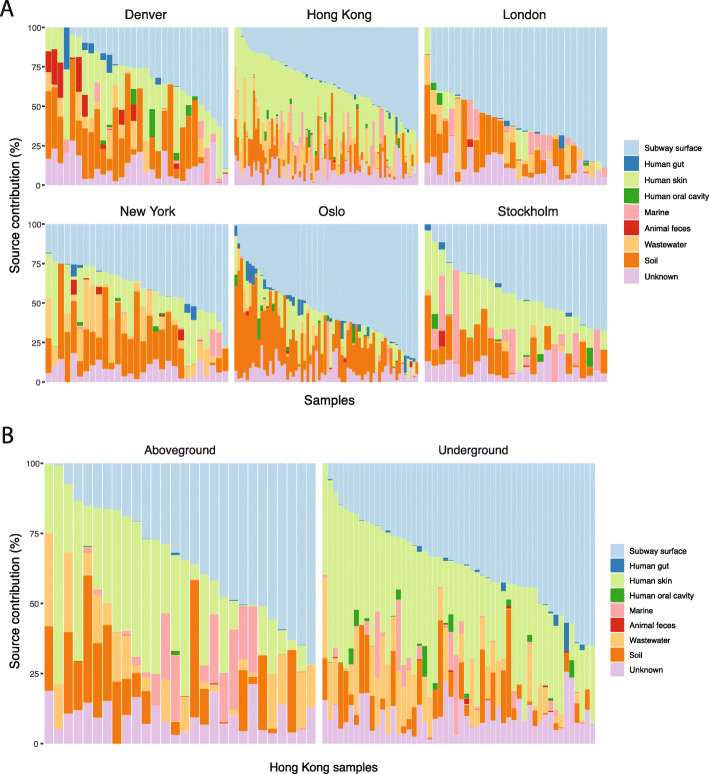
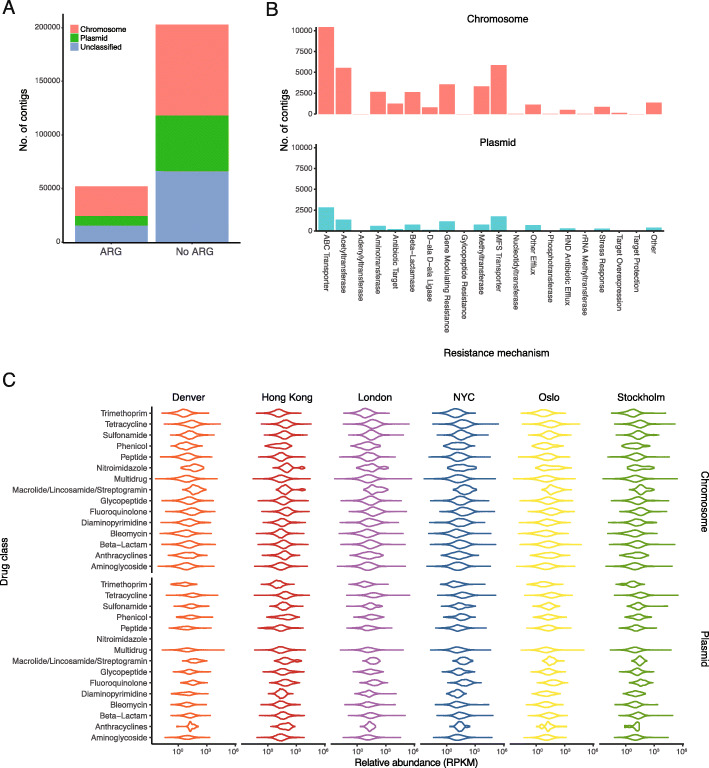
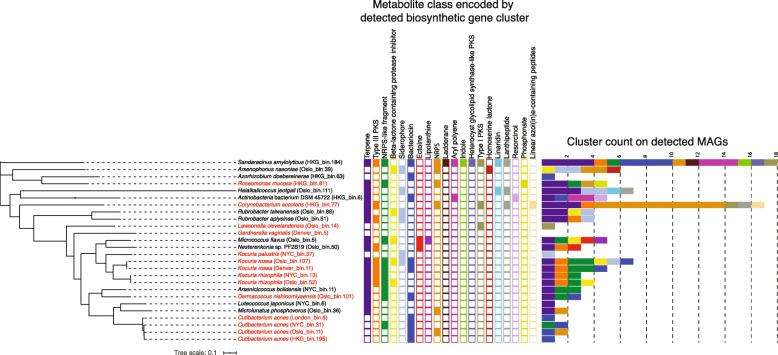
Results: City was the sole factor associated with public transit air microbiome differences, with diverse taxa identified as drivers for geography-associated functional potentials, concomitant with geographical differences in species- and strain-level inferred growth profiles. Related bacterial strains differed among cities in genes encoding resistance, transposase, and other functions. Sourcetracking estimated that human skin, soil, and wastewater were major presumptive resistome sources of public transit air, and adjacent public transit surfaces may also be considered presumptive sources. Large proportions of detected resistance genes were co-located with mobile genetic elements including plasmids. Biosynthetic gene clusters and city-unique coding sequences were found in the metagenome-assembled genomes.
Conclusions: Overall, geographical specificity transcends multiple aspects of the public transit air microbiome, and future efforts on a global scale are warranted to increase our understanding of factors shaping the microbiome of this unique built environment.
Keywords: Air microbiology; Bioinformatics; High-throughput sequencing; Metagenomics; Microbial ecology; Microbiome.
Conflict of interest statement
CEM is a co-founder and shareholder of Biotia, Inc., and Onegevity Health.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
