

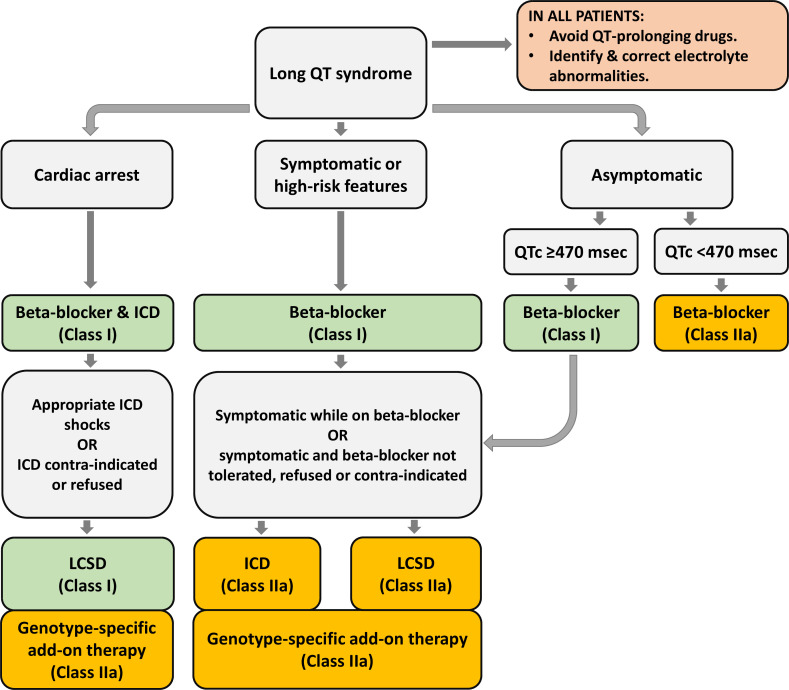
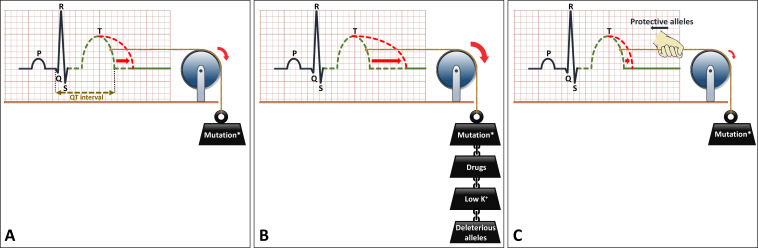
Diagnosis, management and therapeutic strategies for congenital long QT syndrome
- PMID: 34039680
- PMCID: PMC8862104
- DOI: 10.1136/heartjnl-2020-318259
Diagnosis, management and therapeutic strategies for congenital long QT syndrome
Abstract
Congenital long QT syndrome (LQTS) is characterised by heart rate corrected QT interval prolongation and life-threatening arrhythmias, leading to syncope and sudden death. Variations in genes encoding for cardiac ion channels, accessory ion channel subunits or proteins modulating the function of the ion channel have been identified as disease-causing mutations in up to 75% of all LQTS cases. Based on the underlying genetic defect, LQTS has been subdivided into different subtypes. Growing insights into the genetic background and pathophysiology of LQTS has led to the identification of genotype-phenotype relationships for the most common genetic subtypes, the recognition of genetic and non-genetic modifiers of phenotype, optimisation of risk stratification algorithms and the discovery of gene-specific therapies in LQTS. Nevertheless, despite these great advancements in the LQTS field, large gaps in knowledge still exist. For example, up to 25% of LQTS cases still remain genotype elusive, which hampers proper identification of family members at risk, and it is still largely unknown what determines the large variability in disease severity, where even within one family an identical mutation causes malignant arrhythmias in some carriers, while in other carriers, the disease is clinically silent. In this review, we summarise the current evidence available on the diagnosis, clinical management and therapeutic strategies in LQTS. We also discuss new scientific developments and areas of research, which are expected to increase our understanding of the complex genetic architecture in genotype-negative patients, lead to improved risk stratification in asymptomatic mutation carriers and more targeted (gene-specific and even mutation-specific) therapies.
Keywords: genetic services; tachycardia; ventricular; ventricular fibrillation.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
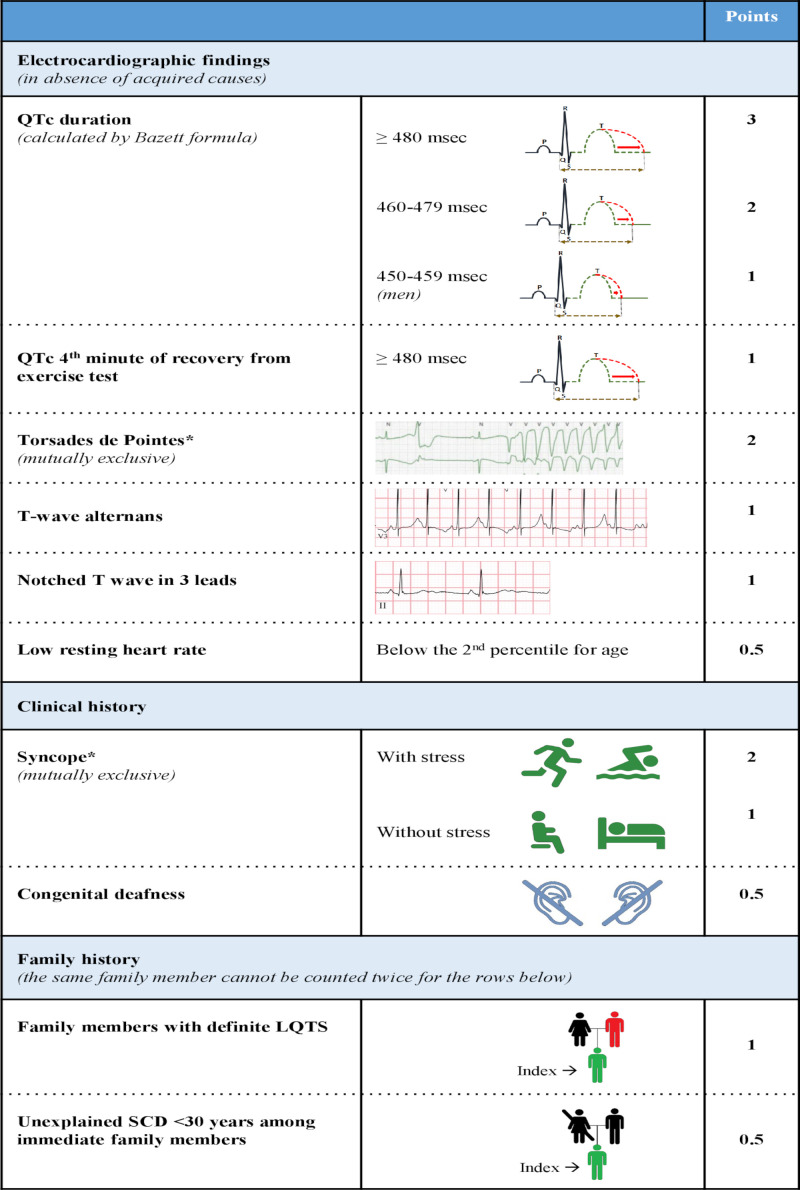
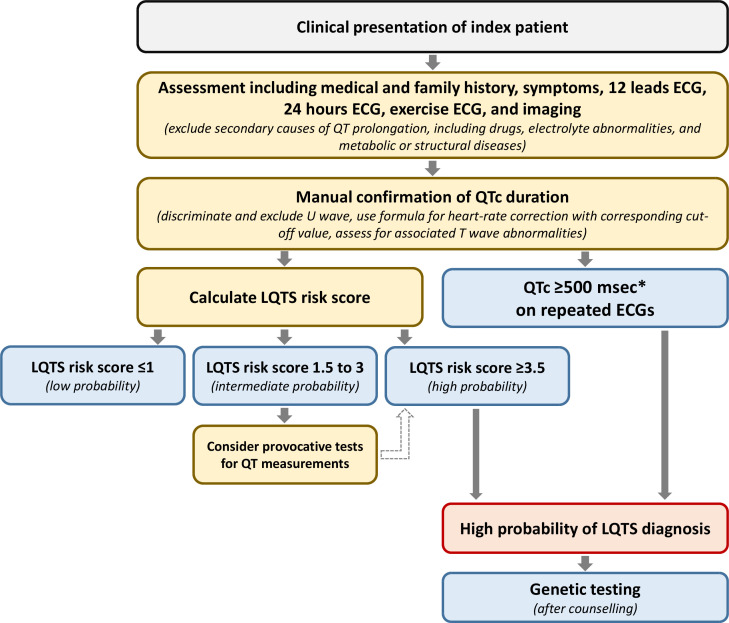
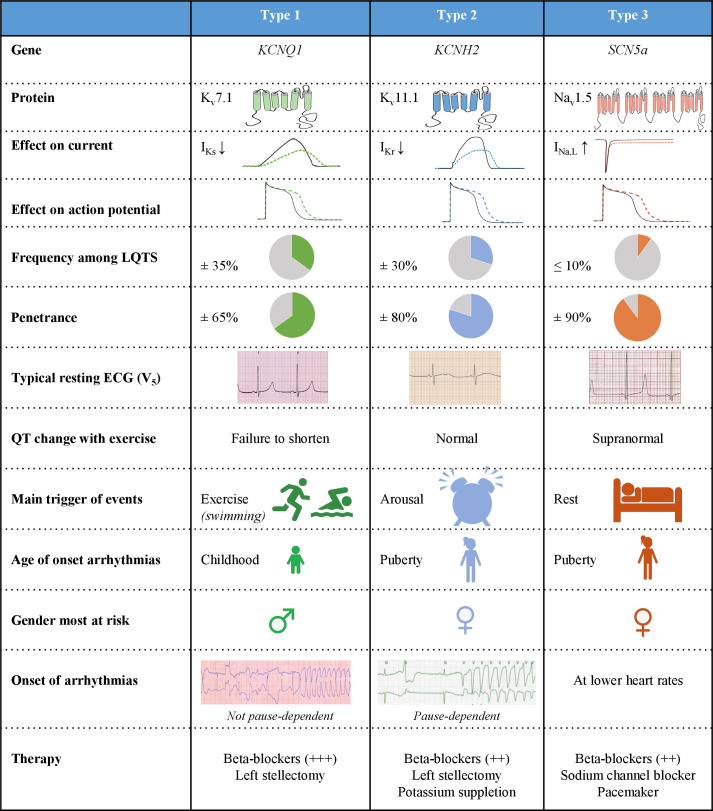
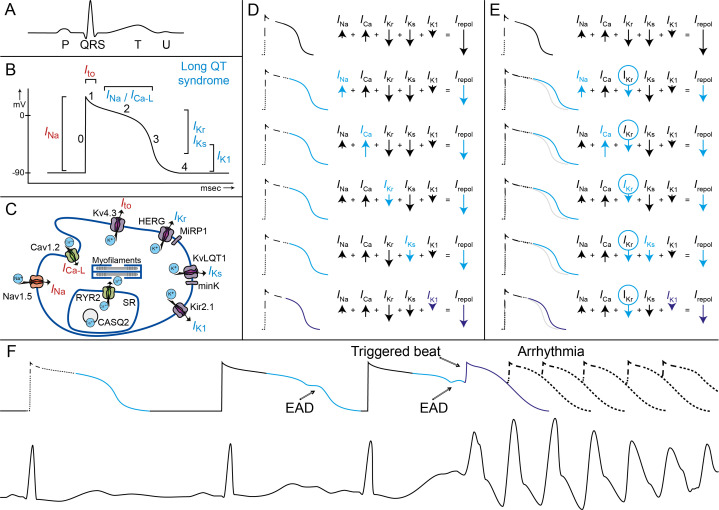
References
-
- PJ: S. 1970-2020: 50 years of research on the long QT syndrome-from almost zero knowledge to precision medicine. Eur Heart J 2020. - PubMed
-
- Priori SG, Wilde AA, Horie M. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;10:1932–63. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources