

A human respiratory tract-associated bacterium with an extremely small genome
- PMID: 34040152
- PMCID: PMC8155191
- DOI: 10.1038/s42003-021-02162-6
A human respiratory tract-associated bacterium with an extremely small genome
Abstract
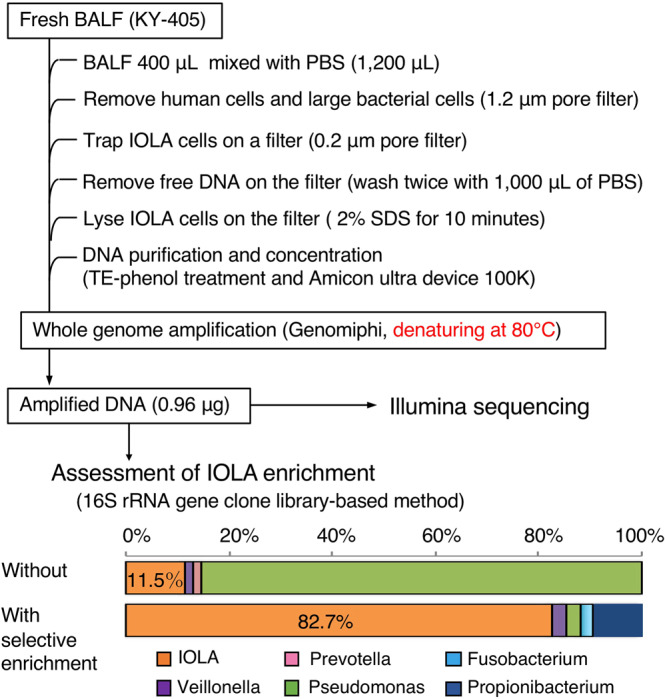
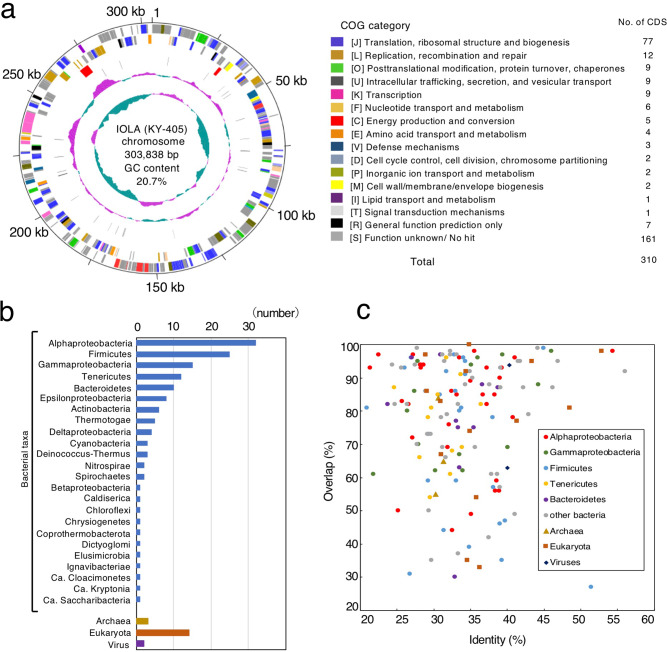
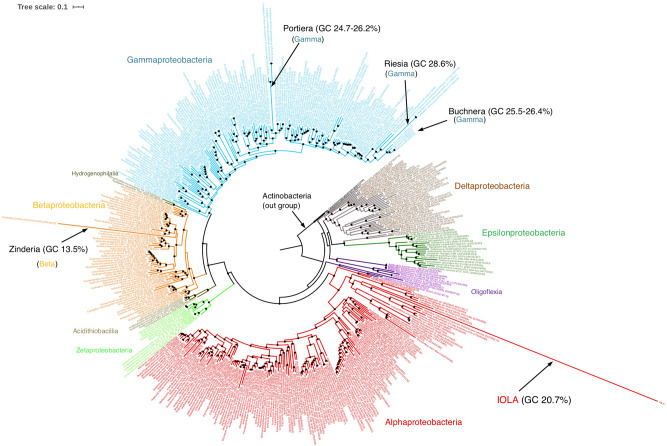
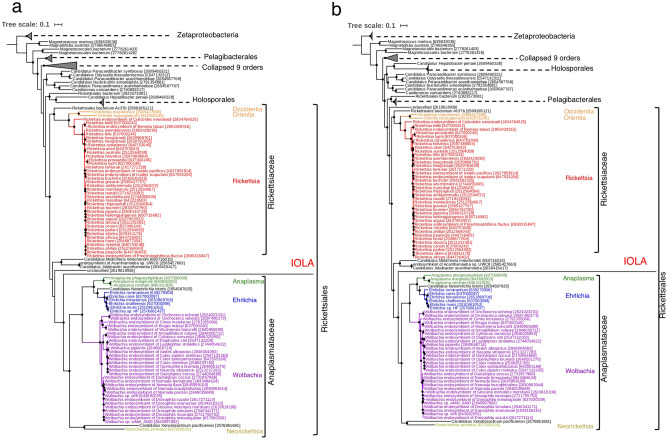
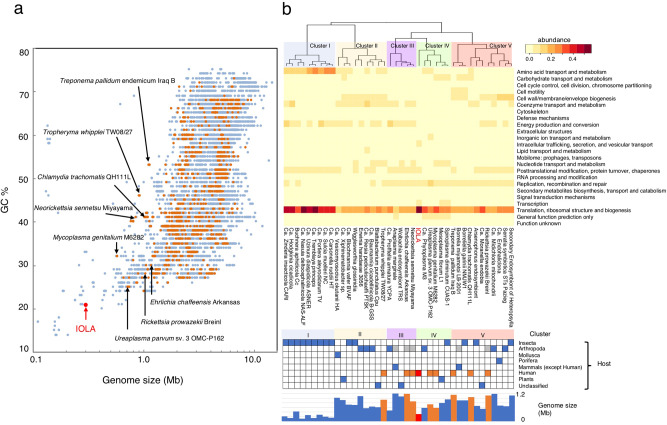
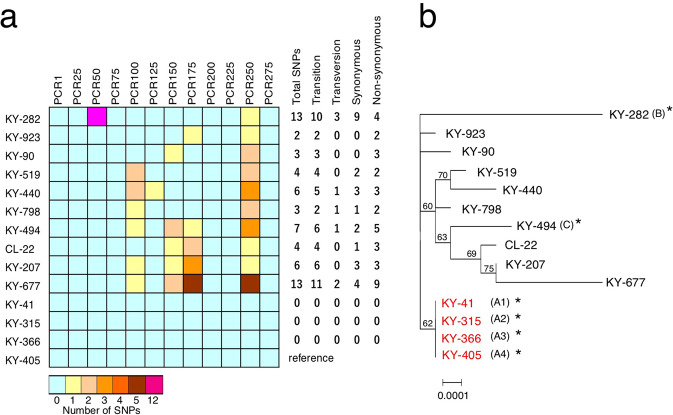
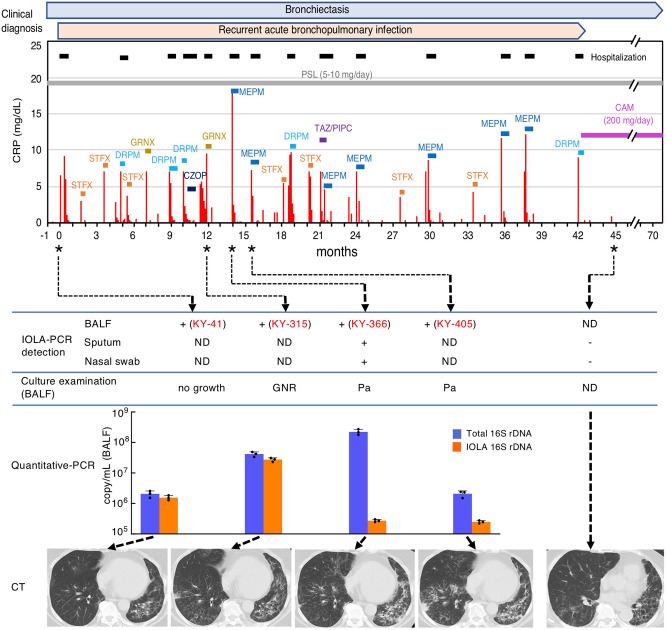
Recent advances in culture-independent microbiological analyses have greatly expanded our understanding of the diversity of unculturable microbes. However, human pathogenic bacteria differing significantly from known taxa have rarely been discovered. Here, we present the complete genome sequence of an uncultured bacterium detected in human respiratory tract named IOLA, which was determined by developing a protocol to selectively amplify extremely AT-rich genomes. The IOLA genome is 303,838 bp in size with a 20.7% GC content, making it the smallest and most AT-rich genome among known human-associated bacterial genomes to our best knowledge and comparable to those of insect endosymbionts. While IOLA belongs to order Rickettsiales (mostly intracellular parasites), the gene content suggests an epicellular parasitic lifestyle. Surveillance of clinical samples provides evidence that IOLA can be predominantly detected in patients with respiratory bacterial infections and can persist for at least 15 months in the respiratory tract, suggesting that IOLA is a human respiratory tract-associated bacterium.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Surveillance and phylogenetic analysis of a pathogenic bacterium candidate in nasal discharge from children.Microbiol Spectr. 2024 Jul 2;12(7):e0056624. doi: 10.1128/spectrum.00566-24. Epub 2024 May 24. Microbiol Spectr. 2024. PMID: 38785433 Free PMC article.
-
An unclassified microorganism: novel pathogen candidate lurking in human airways.PLoS One. 2014 Jul 31;9(7):e103646. doi: 10.1371/journal.pone.0103646. eCollection 2014. PLoS One. 2014. PMID: 25080337 Free PMC article.
-
The evolutionary origin of host association in the Rickettsiales.Nat Microbiol. 2022 Aug;7(8):1189-1199. doi: 10.1038/s41564-022-01169-x. Epub 2022 Jul 7. Nat Microbiol. 2022. PMID: 35798888 Free PMC article.
-
Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle.Nat Rev Microbiol. 2021 Jun;19(6):375-390. doi: 10.1038/s41579-020-00507-2. Epub 2021 Feb 9. Nat Rev Microbiol. 2021. PMID: 33564174 Review.
-
The evolution of bacterial DNA base composition.J Exp Zool B Mol Dev Evol. 2014 Nov;322(7):517-28. doi: 10.1002/jez.b.22565. Epub 2014 Mar 9. J Exp Zool B Mol Dev Evol. 2014. PMID: 24610535 Review.
Cited by
-
Surveillance and phylogenetic analysis of a pathogenic bacterium candidate in nasal discharge from children.Microbiol Spectr. 2024 Jul 2;12(7):e0056624. doi: 10.1128/spectrum.00566-24. Epub 2024 May 24. Microbiol Spectr. 2024. PMID: 38785433 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
