

UBA2 variants underlie a recognizable syndrome with variable aplasia cutis congenita and ectrodactyly
- PMID: 34040189
- PMCID: PMC8463496
- DOI: 10.1038/s41436-021-01182-1
UBA2 variants underlie a recognizable syndrome with variable aplasia cutis congenita and ectrodactyly
Abstract
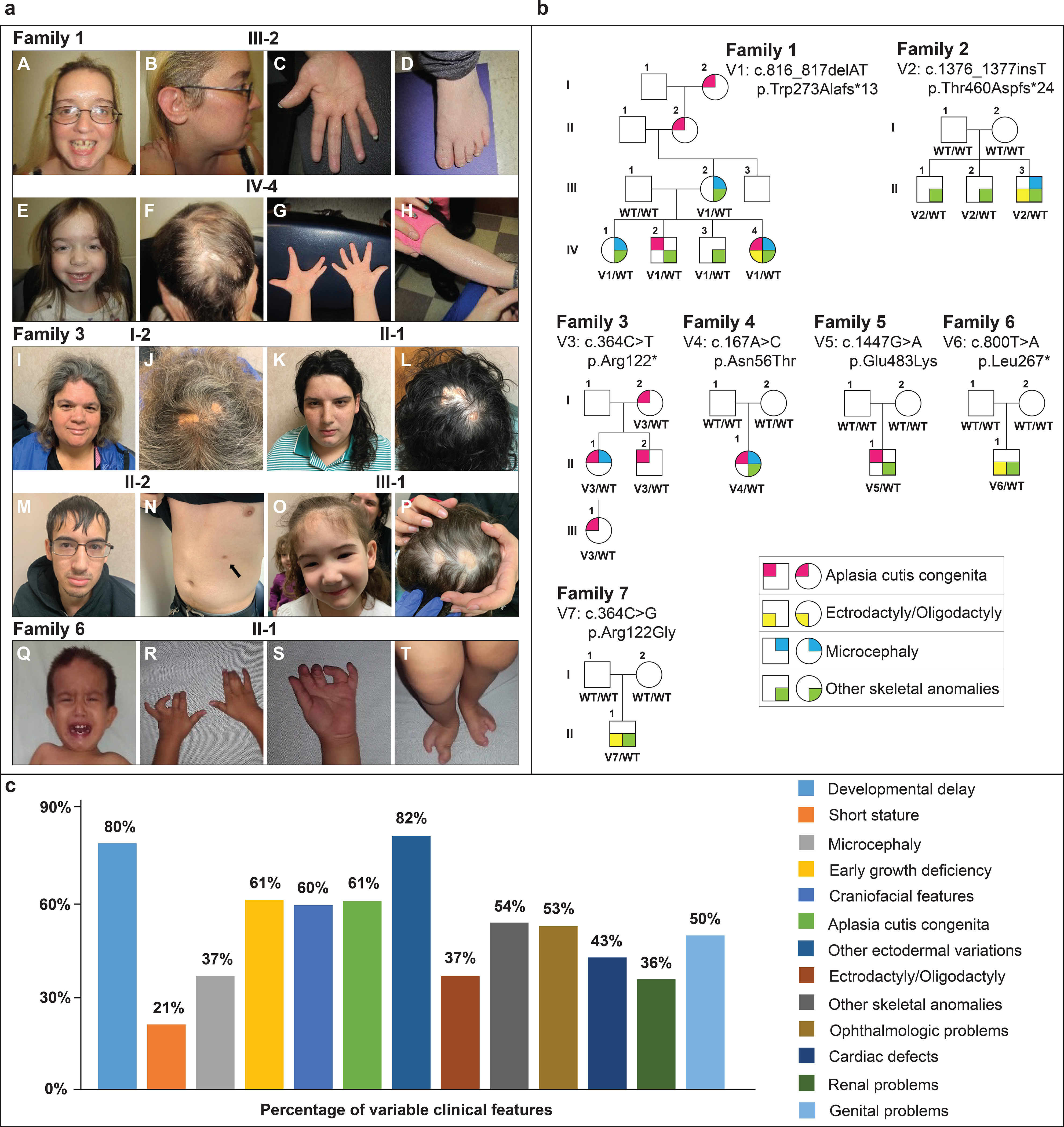
Purpose: The human chromosome 19q13.11 deletion syndrome is associated with a variable phenotype that includes aplasia cutis congenita (ACC) and ectrodactyly as specific features. UBA2 (ubiquitin-like modifier-activating enzyme 2) lies adjacent to the minimal deletion overlap region. We aimed to define the UBA2-related phenotypic spectrum in humans and zebrafish due to sequence variants and to establish the mechanism of disease.
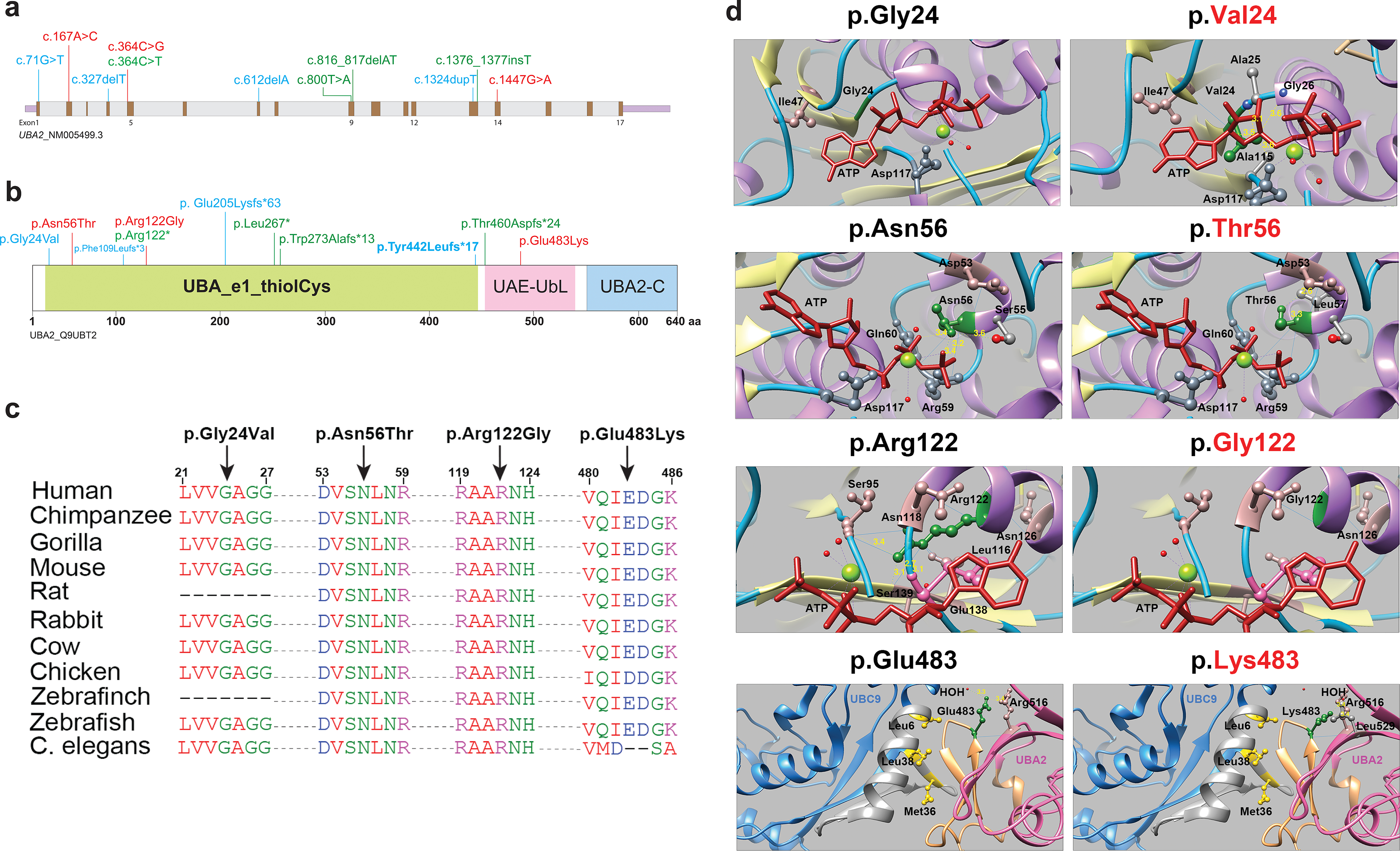
Methods: Exome sequencing was used to detect UBA2 sequence variants in 16 subjects in 7 unrelated families. uba2 loss of function was modeled in zebrafish. Effects of human missense variants were assessed in zebrafish rescue experiments.
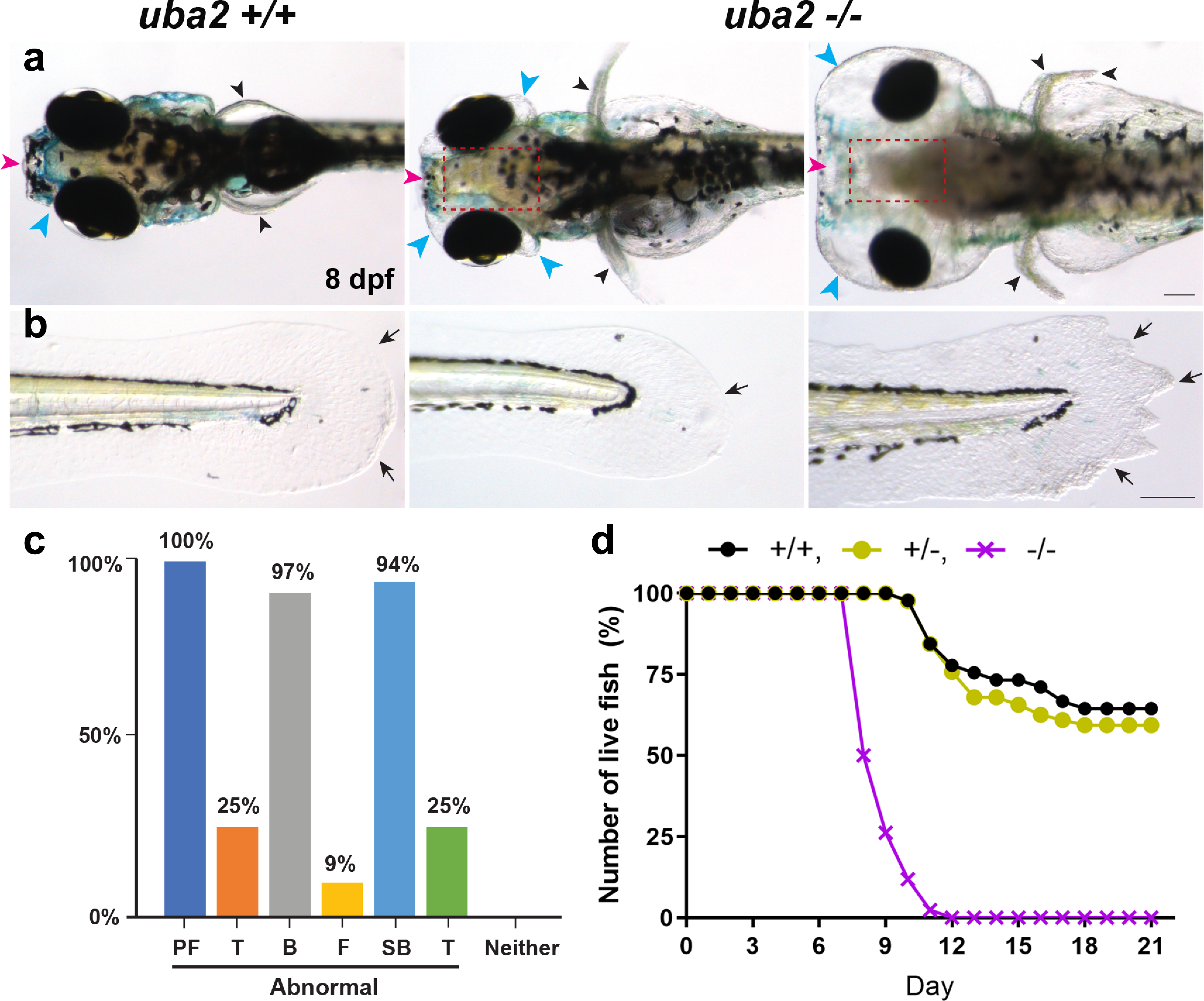
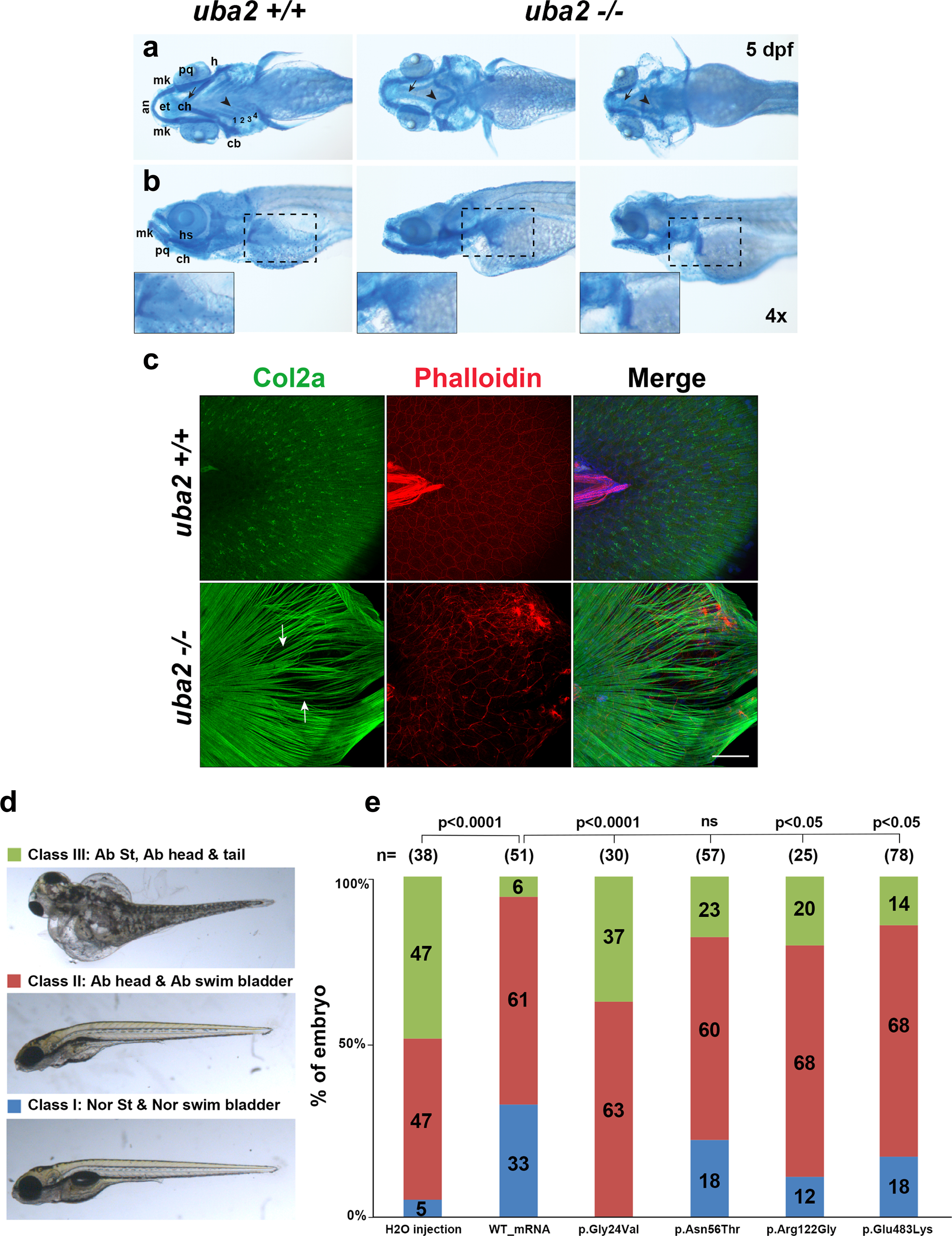
Results: Seven human UBA2 loss-of-function and missense sequence variants were detected. UBA2-phenotypes included ACC, ectrodactyly, neurodevelopmental abnormalities, ectodermal, skeletal, craniofacial, cardiac, renal, and genital anomalies. uba2 was expressed in zebrafish eye, brain, and pectoral fins; uba2-null fish showed deficient growth, microcephaly, microphthalmia, mandibular hypoplasia, and abnormal fins. uba2-mRNAs with human missense variants failed to rescue nullizygous zebrafish phenotypes.
Conclusion: UBA2 variants cause a recognizable syndrome with a wide phenotypic spectrum. Our data suggest that loss of UBA2 function underlies the human UBA2 monogenic disorder and highlights the importance of SUMOylation in the development of affected tissues.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Declaration of Interests:
RES, IMW, MJGS, LR and JJ are employees of GeneDx, Inc., Gaithersburg, MD. The other authors declare no competing interests.
Figures
References
-
- Chowdhury S, Bandholz AM, Parkash S, et al.Phenotypic and molecular characterization of 19q12q13.1 deletions: a report of five patients. Am J Med Genet A. 2014;164A:62–69. - PubMed
-
- Malan V, Raoul O, Firth HV, et al.19q13.11 deletion syndrome: a novel clinically recognisable genetic condition identified by array comparative genomic hybridisation. J Med Genet. 2009;46:635–640. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
