

SNORD116 and growth hormone therapy impact IGFBP7 in Prader-Willi syndrome
- PMID: 34040195
- PMCID: PMC8460435
- DOI: 10.1038/s41436-021-01185-y
SNORD116 and growth hormone therapy impact IGFBP7 in Prader-Willi syndrome
Abstract
Purpose: Prader-Willi syndrome (PWS) is a neurodevelopmental disorder with hypothalamic dysfunction due to deficiency of imprinted genes located on the 15q11-q13 chromosome. Among them, the SNORD116 gene appears critical for the expression of the PWS phenotype. We aimed to clarify the role of SNORD116 in cellular and animal models with regard to growth hormone therapy (GHT), the main approved treatment for PWS.
Methods: We collected serum and induced pluripotent stem cells (iPSCs) from GH-treated PWS patients to differentiate into dopaminergic neurons, and in parallel used a Snord116 knockout mouse model. We analyzed the expression of factors potentially linked to GH responsiveness.
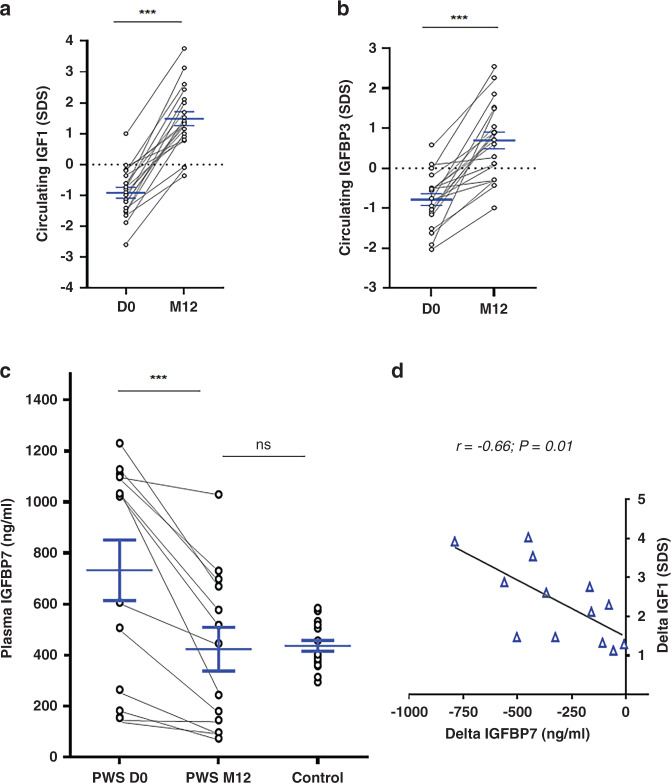
Results: We found elevated levels of circulating IGFBP7 in naive PWS patients, with IGFBP7 levels normalizing under GHT. We found elevated IGFBP7 levels in the brains of Snord116 knockout mice and in iPSC-derived neurons from a SNORD116-deleted PWS patient. High circulating levels of IGFBP7 in PWS patients may result from both increased IGFBP7 expression and decreased IGFBP7 cleavage, by downregulation of the proconvertase PC1.
Conclusion: SNORD116 deletion affects IGFBP7 levels, while IGFBP7 decreases under GHT in PWS patients. Modulation of the IGFBP7 level, which interacts with IGF1, has implications in the pathophysiology and management of PWS under GHT.
Trial registration: ClinicalTrials.gov NCT01298180.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
RNAi Knockdown of EHMT2 in Maternal Expression of Prader-Willi Syndrome Genes.Genes (Basel). 2024 Oct 24;15(11):1366. doi: 10.3390/genes15111366. Genes (Basel). 2024. PMID: 39596566 Free PMC article.
-
Reference Genes across Nine Brain Areas of Wild Type and Prader-Willi Syndrome Mice: Assessing Differences in Igfbp7, Pcsk1, Nhlh2 and Nlgn3 Expression.Int J Mol Sci. 2022 Aug 5;23(15):8729. doi: 10.3390/ijms23158729. Int J Mol Sci. 2022. PMID: 35955861 Free PMC article.
-
Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome.J Clin Invest. 2017 Jan 3;127(1):293-305. doi: 10.1172/JCI88648. Epub 2016 Dec 12. J Clin Invest. 2017. PMID: 27941249 Free PMC article.
-
Cognitive deficits in the Snord116 deletion mouse model for Prader-Willi syndrome.Neurobiol Learn Mem. 2019 Nov;165:106874. doi: 10.1016/j.nlm.2018.05.011. Epub 2018 May 23. Neurobiol Learn Mem. 2019. PMID: 29800646 Free PMC article. Review.
-
Epigenetic therapy of Prader-Willi syndrome.Transl Res. 2019 Jun;208:105-118. doi: 10.1016/j.trsl.2019.02.012. Epub 2019 Mar 5. Transl Res. 2019. PMID: 30904443 Free PMC article. Review.
Cited by
-
Approach to the Patient With Prader-Willi Syndrome.J Clin Endocrinol Metab. 2022 May 17;107(6):1698-1705. doi: 10.1210/clinem/dgac082. J Clin Endocrinol Metab. 2022. PMID: 35150573 Free PMC article.
-
Linking oxytocin and arginine vasopressin signaling abnormalities to social behavior impairments in Prader-Willi syndrome.Neurosci Biobehav Rev. 2022 Nov;142:104870. doi: 10.1016/j.neubiorev.2022.104870. Epub 2022 Sep 13. Neurosci Biobehav Rev. 2022. PMID: 36113782 Free PMC article. Review.
-
Hormonal Imbalances in Prader-Willi and Schaaf-Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals.Int J Mol Sci. 2023 Aug 23;24(17):13109. doi: 10.3390/ijms241713109. Int J Mol Sci. 2023. PMID: 37685915 Free PMC article. Review.
-
Differential DNA methylation in iPSC-derived dopaminergic neurons: a step forward on the role of SNORD116 microdeletion in the pathophysiology of addictive behavior in Prader-Willi syndrome.Mol Psychiatry. 2024 Sep;29(9):2742-2752. doi: 10.1038/s41380-024-02542-4. Epub 2024 Apr 2. Mol Psychiatry. 2024. PMID: 38561465
-
Common and Uncommon Mouse Models of Growth Hormone Deficiency.Endocr Rev. 2024 Nov 22;45(6):818-842. doi: 10.1210/endrev/bnae017. Endocr Rev. 2024. PMID: 38853618 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
