

Whole-Genome Analysis of Mycobacterium avium subsp. paratuberculosis IS 900 Insertions Reveals Strain Type- Specific Modalities
- PMID: 34040595
- PMCID: PMC8141618
- DOI: 10.3389/fmicb.2021.660002
Whole-Genome Analysis of Mycobacterium avium subsp. paratuberculosis IS 900 Insertions Reveals Strain Type- Specific Modalities
Abstract
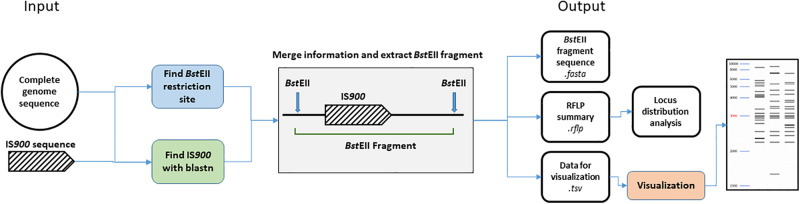
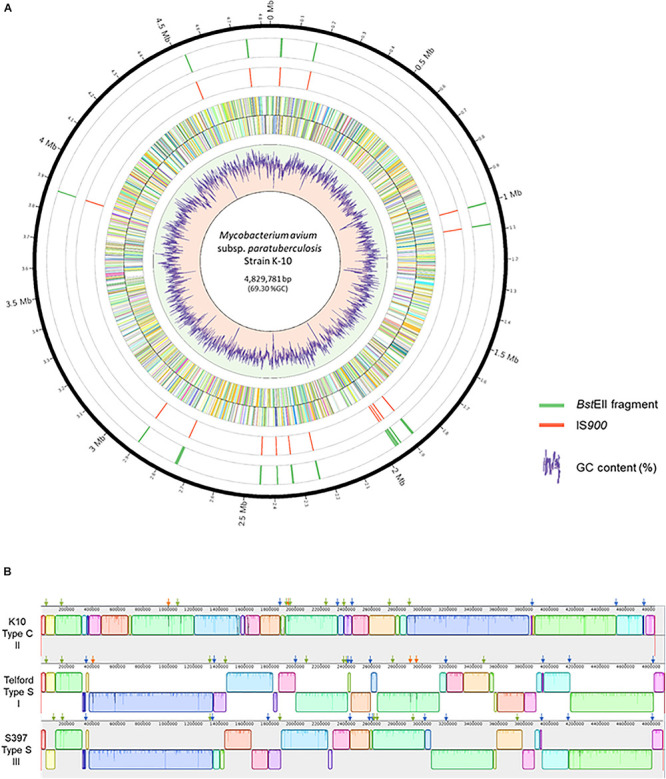
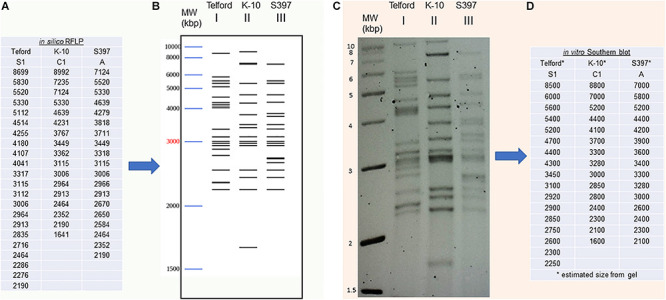
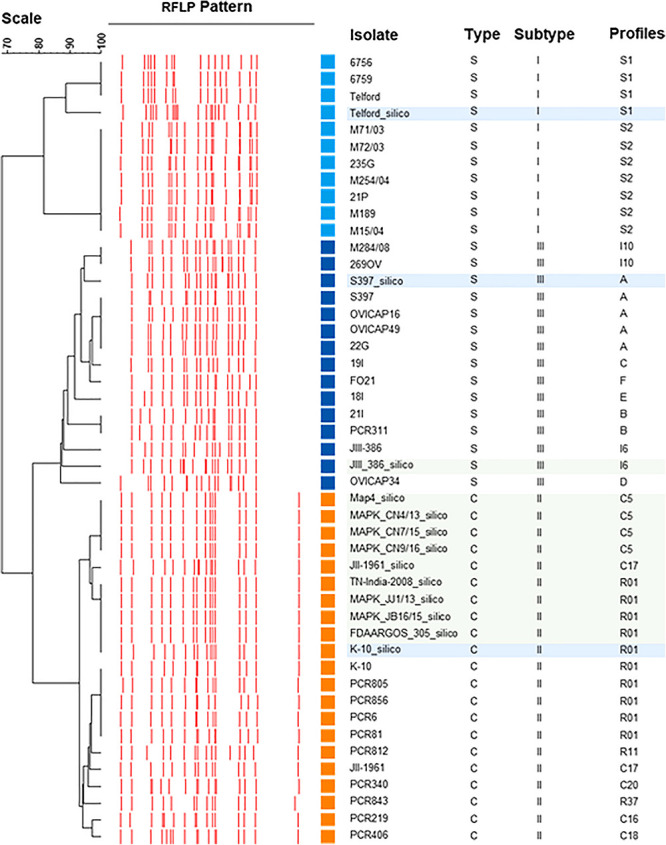
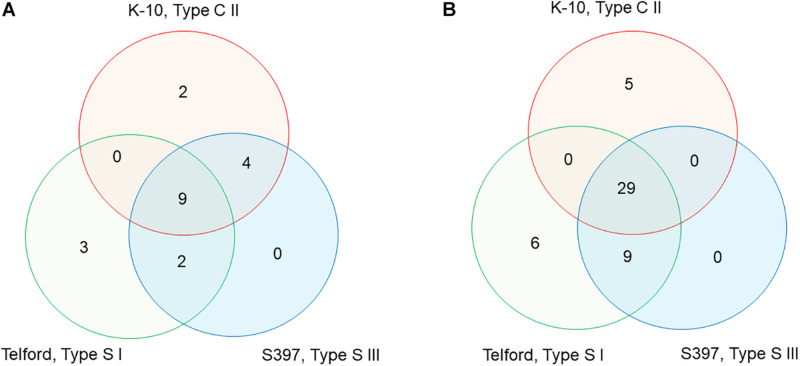
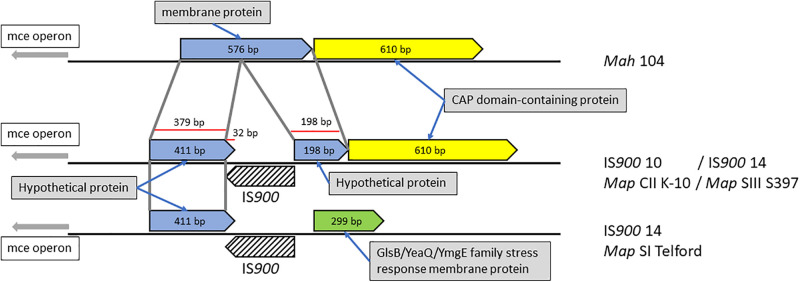
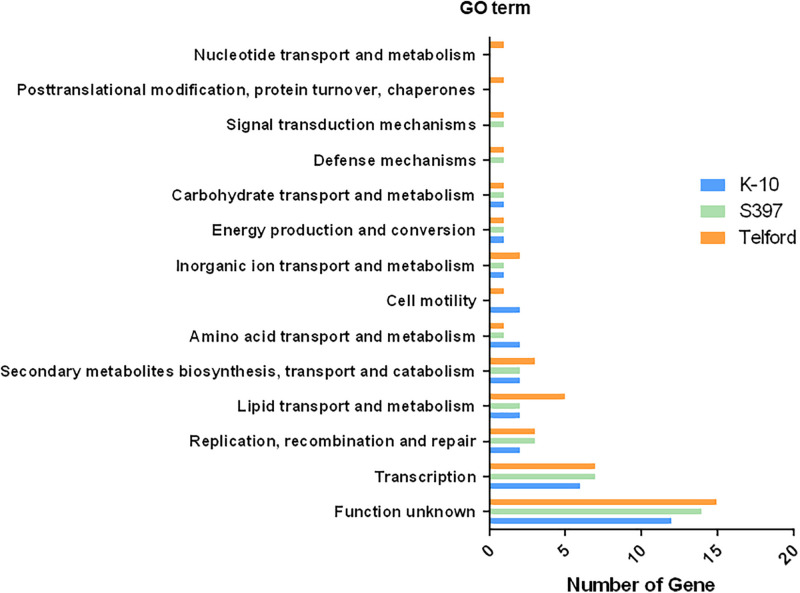
Mycobacterium avium subsp. paratuberculosis (Map) is the etiological agent of Johne's disease in ruminants. The IS900 insertion sequence (IS) has been used widely as an epidemiological marker and target for PCR diagnosis. Updated DNA sequencing technologies have led to a rapid increase in available Map genomes, which makes it possible to analyze the distribution of IS900 in this slow-growing bacterium. The objective of this study is to characterize the distribution of the IS900 element and how it affects genomic evolution and gene function of Map. A secondary goal is to develop automated in silico restriction fragment length polymorphism (RFLP) analysis using IS900. Complete genomes from the major phylogenetic lineages known as C-type and S-type (including subtypes I and III), were chosen to represent the genetic diversity of Map. IS900 elements were located in these genomes using BLAST software and the relevant fragments extracted. An in silico RFLP analysis using the BstEII restriction site was performed to obtain exact sizes of the DNA fragments carrying a copy of IS900 and the resulting RFLP profiles were analyzed and compared by digital visualization of the separated restriction fragments. The program developed for this study allowed automated localization of IS900 sequences to identify their position within each genome along with the exact number of copies per genome. The number of IS900 copies ranged from 16 in the C-type isolate to 22 in the S-type subtype I isolate. A loci-by-loci sequence alignment of all IS900 copies within the three genomes revealed new sequence polymorphisms that define three sequevars distinguishing the subtypes. Nine IS900 insertion site locations were conserved across all genomes studied while smaller subsets were unique to a particular lineage. Preferential insertion motif sequences were identified for IS900 along with genes bordering all IS900 insertions. Rarely did IS900 insert within coding sequences as only three genes were disrupted in this way. This study makes it possible to automate IS900 distribution in Map genomes to enrich knowledge on the distribution dynamics of this IS for epidemiological purposes, for understanding Map evolution and for studying the biological implications of IS900 insertions.
Keywords: Mycobacterium avium subsp. paratuberculosis; RFLP; complete genome; evolution; insertion sequence IS900.
Copyright © 2021 Conde, Price-Carter, Cochard, Branger, Stevenson, Whittington, Bannantine and Biet.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Characterization of IS900 loci in Mycobacterium avium subsp. paratuberculosis and development of multiplex PCR typing.Microbiology (Reading). 2000 Sep;146 ( Pt 9):2185-2197. doi: 10.1099/00221287-146-9-2185. Microbiology (Reading). 2000. PMID: 10974106
-
High genetic diversity among Mycobacterium avium subsp. paratuberculosis strains from German cattle herds shown by combination of IS900 restriction fragment length polymorphism analysis and mycobacterial interspersed repetitive unit-variable-number tandem-repeat typing.J Clin Microbiol. 2008 Mar;46(3):972-81. doi: 10.1128/JCM.01801-07. Epub 2008 Jan 3. J Clin Microbiol. 2008. PMID: 18174306 Free PMC article.
-
Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis: IS900 restriction fragment length polymorphism and IS1311 polymorphism analyses of isolates from animals and a human in Australia.J Clin Microbiol. 2000 Sep;38(9):3240-8. doi: 10.1128/JCM.38.9.3240-3248.2000. J Clin Microbiol. 2000. PMID: 10970365 Free PMC article.
-
Strain diversity within Mycobacterium avium subspecies paratuberculosis--a review.Indian J Exp Biol. 2010 Jan;48(1):7-16. Indian J Exp Biol. 2010. PMID: 20358861 Review.
-
Current understanding of the genetic diversity of Mycobacterium avium subsp. paratuberculosis.Microbes Infect. 2006 Apr;8(5):1406-18. doi: 10.1016/j.micinf.2005.12.003. Epub 2006 Jan 26. Microbes Infect. 2006. PMID: 16697677 Review.
Cited by
-
Evidence of Mycobacterium bovis DNA in shared water sources at livestock-wildlife-human interfaces in KwaZulu-Natal, South Africa.Front Vet Sci. 2025 Feb 28;12:1483162. doi: 10.3389/fvets.2025.1483162. eCollection 2025. Front Vet Sci. 2025. PMID: 40093619 Free PMC article.
-
Improved DNA Amplification of the Hallmark IS900 Element in Mycobacterium avium subsp. paratuberculosis: a Reexamination Based on Whole-Genome Sequence Analysis.Appl Environ Microbiol. 2023 Feb 28;89(2):e0168222. doi: 10.1128/aem.01682-22. Epub 2023 Jan 31. Appl Environ Microbiol. 2023. PMID: 36719222 Free PMC article.
-
Bovine Immunity and Vitamin D3: An Emerging Association in Johne's Disease.Microorganisms. 2022 Sep 18;10(9):1865. doi: 10.3390/microorganisms10091865. Microorganisms. 2022. PMID: 36144467 Free PMC article. Review.
-
Mycobacterium avium subsp. paratuberculosis Virulence: A Review.Microorganisms. 2021 Dec 19;9(12):2623. doi: 10.3390/microorganisms9122623. Microorganisms. 2021. PMID: 34946224 Free PMC article. Review.
-
Investigating in vivo Mycobacterium avium subsp. paratuberculosis microevolution and mixed strain infections.Microbiol Spectr. 2023 Aug 16;11(5):e0171623. doi: 10.1128/spectrum.01716-23. Online ahead of print. Microbiol Spectr. 2023. PMID: 37584606 Free PMC article.
References
-
- Bailey T. L., Elkan C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell Syst. Mol. Biol. 2 28–36. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials