

The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy
- PMID: 34041026
- PMCID: PMC8141735
- DOI: 10.3389/fonc.2021.656229
The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy
Abstract
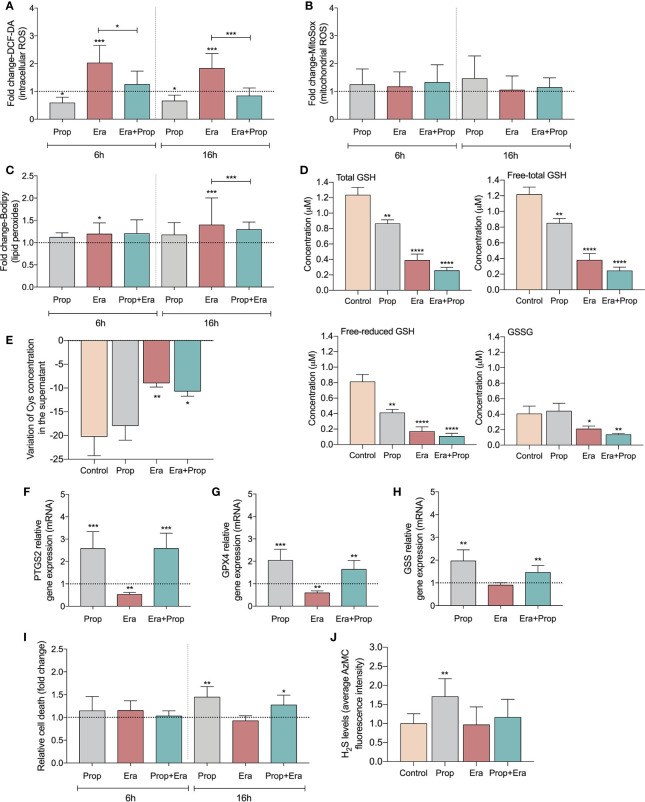
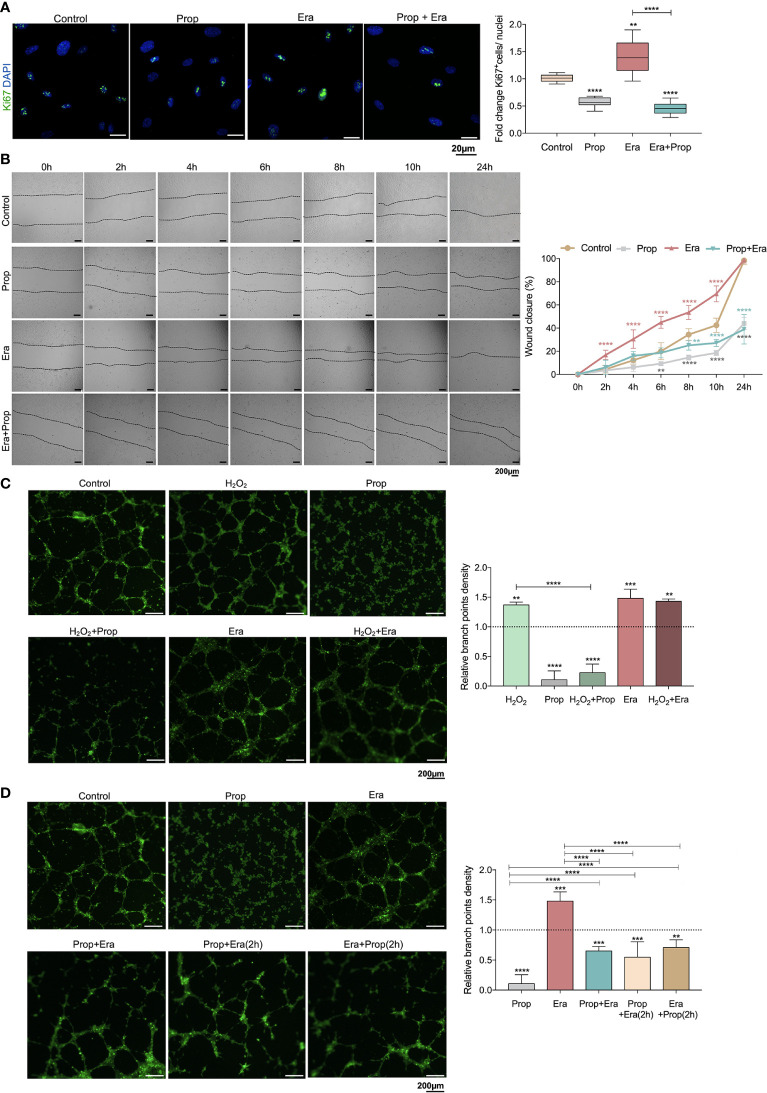
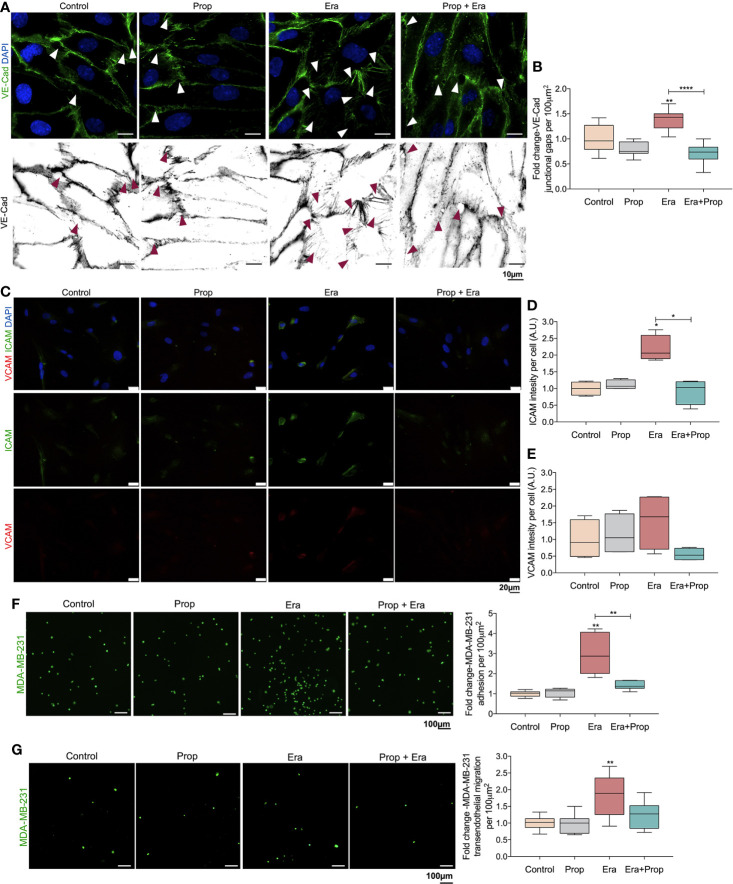
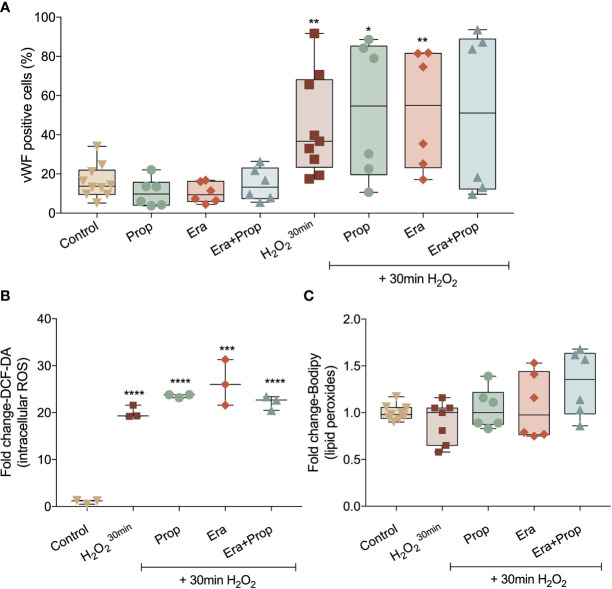
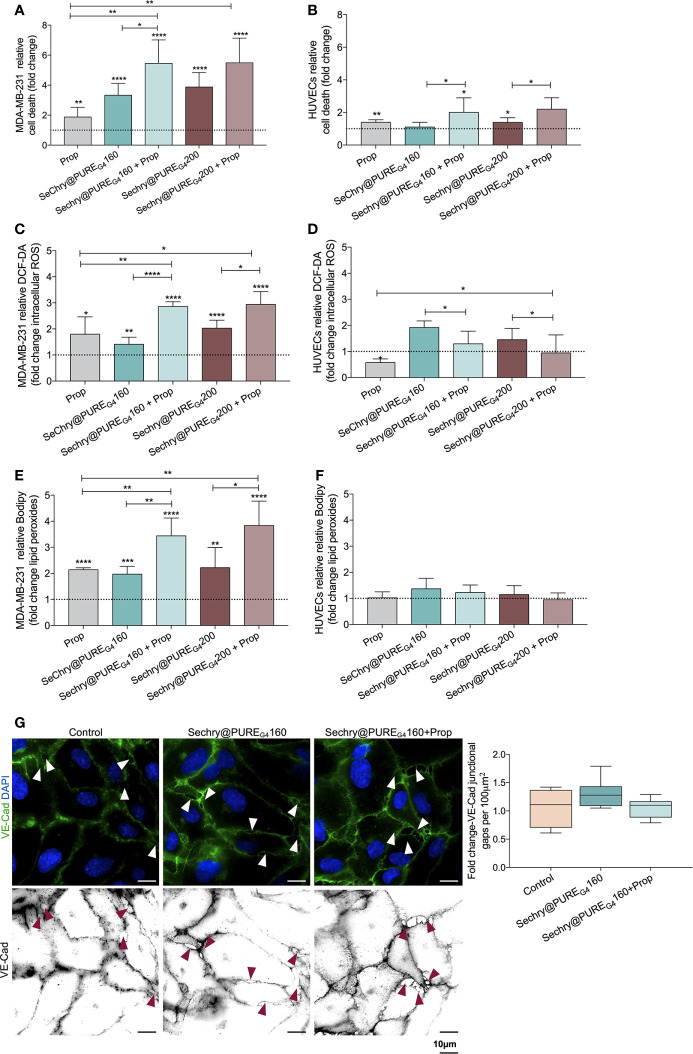
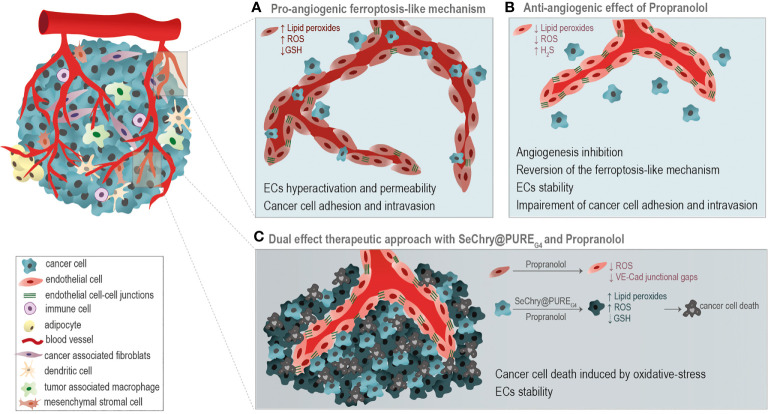
The activation of endothelial cells (ECs) is a crucial step on the road map of tumor angiogenesis and expanding evidence indicates that a pro-oxidant tumor microenvironment, conditioned by cancer metabolic rewiring, is a relevant controller of this process. Herein, we investigated the contribution of oxidative stress-induced ferroptosis to ECs activation. Moreover, we also addressed the anti-angiogenic effect of Propranolol. We observed that a ferroptosis-like mechanism, induced by xCT inhibition with Erastin, at a non-lethal level, promoted features of ECs activation, such as proliferation, migration and vessel-like structures formation, concomitantly with the depletion of reduced glutathione (GSH) and increased levels of oxidative stress and lipid peroxides. Additionally, this ferroptosis-like mechanism promoted vascular endothelial cadherin (VE-cadherin) junctional gaps and potentiated cancer cell adhesion to ECs and transendothelial migration. Propranolol was able to revert Erastin-dependent activation of ECs and increased levels of hydrogen sulfide (H2S) underlie the mechanism of action of Propranolol. Furthermore, we tested a dual-effect therapy by promoting ECs stability with Propranolol and boosting oxidative stress to induce cancer cell death with a nanoformulation comprising selenium-containing chrysin (SeChry) encapsulated in a fourth generation polyurea dendrimer (SeChry@PUREG4). Our data showed that novel developments in cancer treatment may rely on multi-targeting strategies focusing on nanoformulations for a safer induction of cancer cell death, taking advantage of tumor vasculature stabilization.
Keywords: angiogenesis; endothelial cell hyperactivation; ferroptosis; lipid peroxidation; oxidative stress; polyurea dendrimers; propranolol; tumor vasculature stabilizers.
Copyright © 2021 Lopes-Coelho, Martins, Hipólito, Mendes, Sequeira, Pires, Almeida, Bonifácio, Pereira and Serpa.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium-Chrysin Polyurea Dendrimer Nanoformulation.Nutrients. 2019 Oct 19;11(10):2523. doi: 10.3390/nu11102523. Nutrients. 2019. PMID: 31635026 Free PMC article.
-
ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner.Oncogene. 2017 Oct 5;36(40):5593-5608. doi: 10.1038/onc.2017.146. Epub 2017 May 29. Oncogene. 2017. PMID: 28553953 Free PMC article.
-
Lipid Peroxidation, GSH Depletion, and SLC7A11 Inhibition Are Common Causes of EMT and Ferroptosis in A549 Cells, but Different in Specific Mechanisms.DNA Cell Biol. 2021 Feb;40(2):172-183. doi: 10.1089/dna.2020.5730. Epub 2020 Dec 22. DNA Cell Biol. 2021. PMID: 33351681
-
Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis.Curr Top Microbiol Immunol. 2017;403:143-170. doi: 10.1007/82_2016_508. Curr Top Microbiol Immunol. 2017. PMID: 28204974 Review.
-
Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs.Cancer Lett. 2020 Jul 28;483:127-136. doi: 10.1016/j.canlet.2020.02.015. Epub 2020 Feb 14. Cancer Lett. 2020. PMID: 32067993 Review.
Cited by
-
CXCL8 Promotes Endothelial-to-Mesenchymal Transition of Endothelial Cells and Protects Cells from Erastin-Induced Ferroptosis via CXCR2-Mediated Activation of the NF-κB Signaling Pathway.Pharmaceuticals (Basel). 2023 Aug 25;16(9):1210. doi: 10.3390/ph16091210. Pharmaceuticals (Basel). 2023. PMID: 37765018 Free PMC article.
-
The CBS-H2S axis promotes liver metastasis of colon cancer by upregulating VEGF through AP-1 activation.Br J Cancer. 2022 Apr;126(7):1055-1066. doi: 10.1038/s41416-021-01681-7. Epub 2021 Dec 24. Br J Cancer. 2022. PMID: 34952931 Free PMC article.
-
Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex.Antioxidants (Basel). 2021 Sep 17;10(9):1484. doi: 10.3390/antiox10091484. Antioxidants (Basel). 2021. PMID: 34573115 Free PMC article.
-
Vascular endothelial growth factor A inhibition remodels the transcriptional signature of lipid metabolism in psoriasis non-lesional skin in 12 h ex vivo culture.Skin Health Dis. 2024 Oct 26;4(6):e471. doi: 10.1002/ski2.471. eCollection 2024 Dec. Skin Health Dis. 2024. PMID: 39624732 Free PMC article.
-
Intercellular Adhesion Molecule 1 (ICAM-1): An Inflammatory Regulator with Potential Implications in Ferroptosis and Parkinson's Disease.Cells. 2024 Sep 15;13(18):1554. doi: 10.3390/cells13181554. Cells. 2024. PMID: 39329738 Free PMC article. Review.
References
-
- Hanahan D, Folkman J. Patterns and Emerging Mechanisms of the Angiogenic Switch During Tumorigenesis. Cell (1996) 86:353–64. S0092-8674(00)80108-7 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources