

CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents
- PMID: 34041038
- PMCID: PMC8143439
- DOI: 10.3389/fonc.2021.678559
CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents
Abstract
Cyclin-dependent kinases (CDKs) are proteins pivotal to a wide range of cellular functions, most importantly cell division and transcription, and their dysregulations have been implicated as prominent drivers of tumorigenesis. Besides the well-established role of cell cycle CDKs in cancer, the involvement of transcriptional CDKs has been confirmed more recently. Most cancers overtly employ CDKs that serve as key regulators of transcription (e.g., CDK9) for a continuous production of short-lived gene products that maintain their survival. As such, dysregulation of the CDK9 pathway has been observed in various hematological and solid malignancies, making it a valuable anticancer target. This therapeutic potential has been utilized for the discovery of CDK9 inhibitors, some of which have entered human clinical trials. This review provides a comprehensive discussion on the structure and biology of CDK9, its role in solid and hematological cancers, and an updated review of the available inhibitors currently being investigated in preclinical and clinical settings.
Keywords: CDK9 inhibitors; CDKs; P-TEFb; cancer; transcription.
Copyright © 2021 Anshabo, Milne, Wang and Albrecht.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
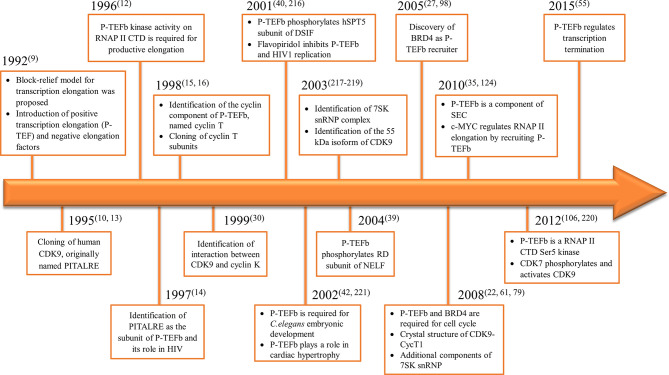
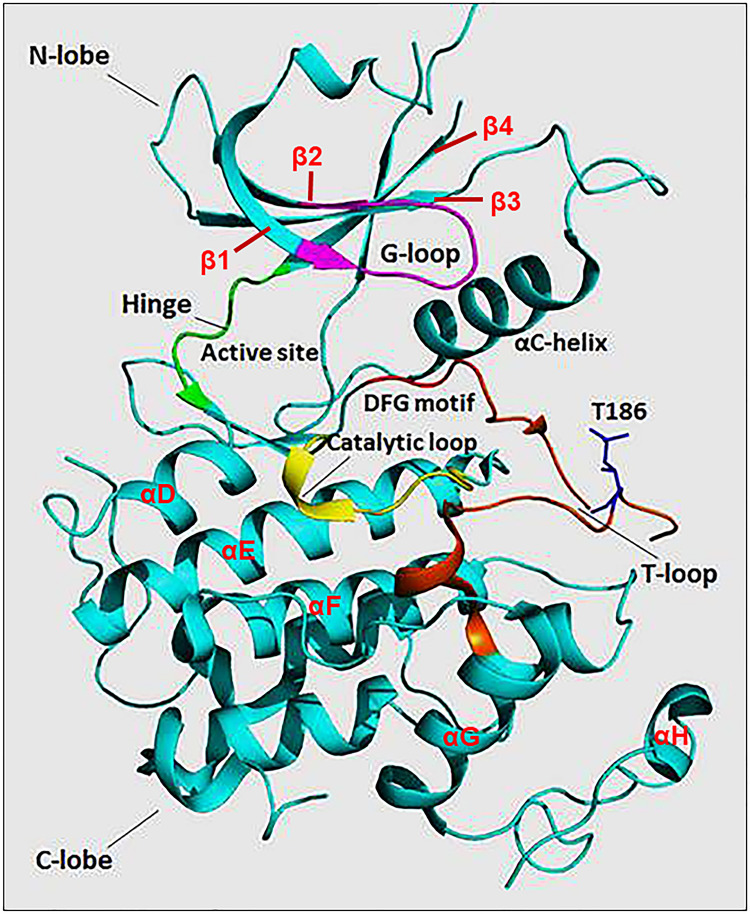


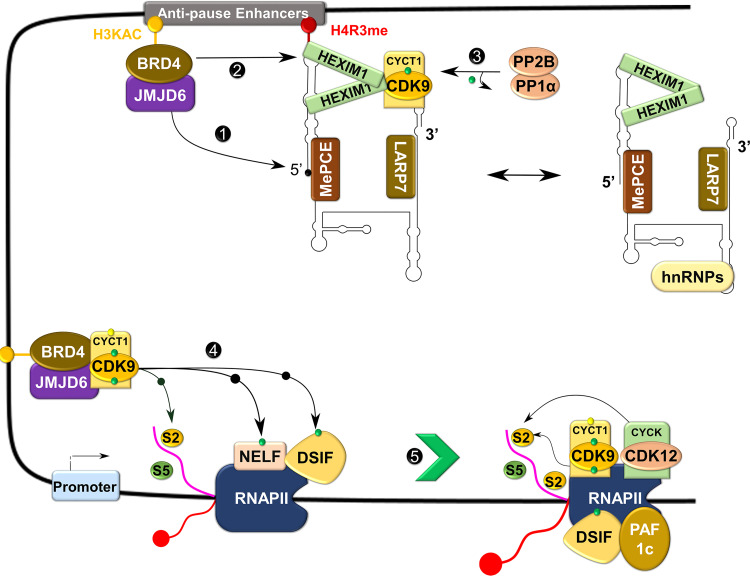
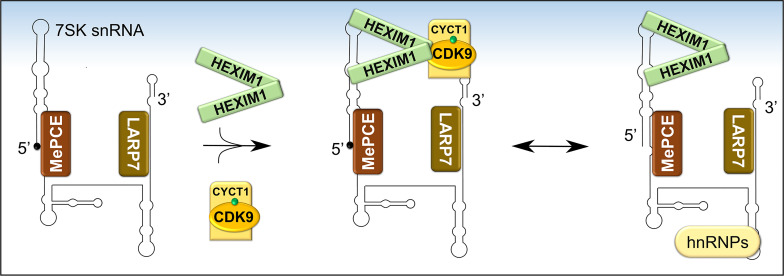
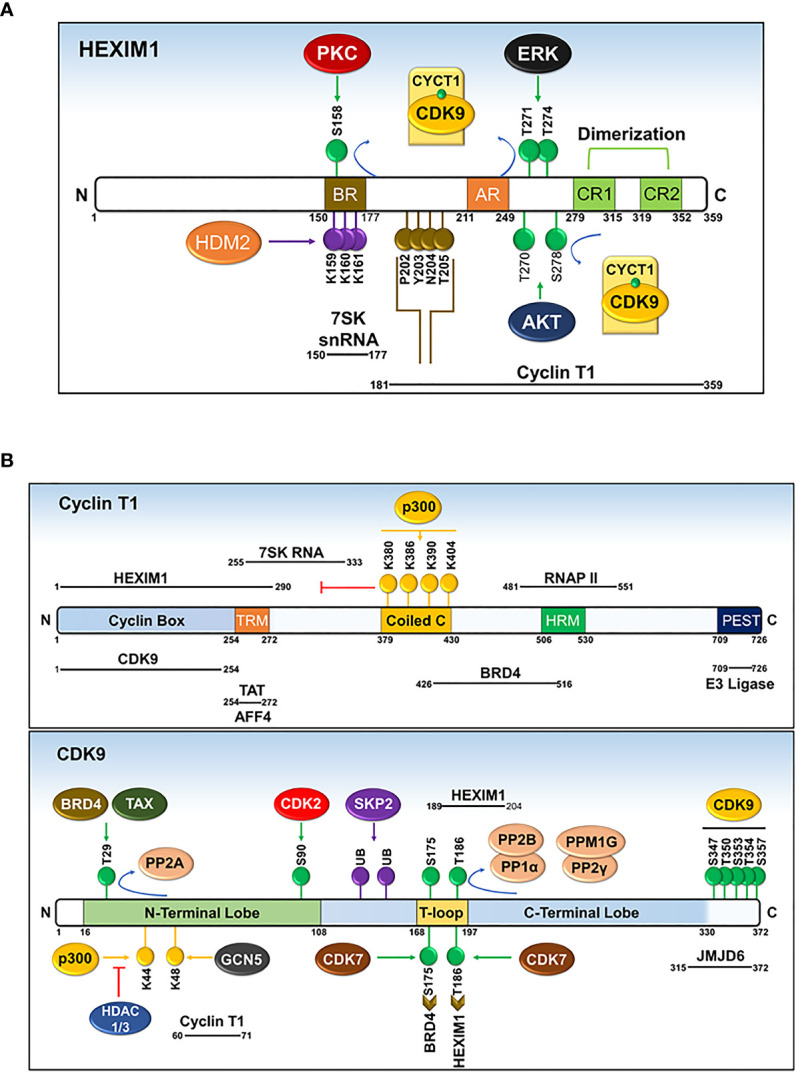

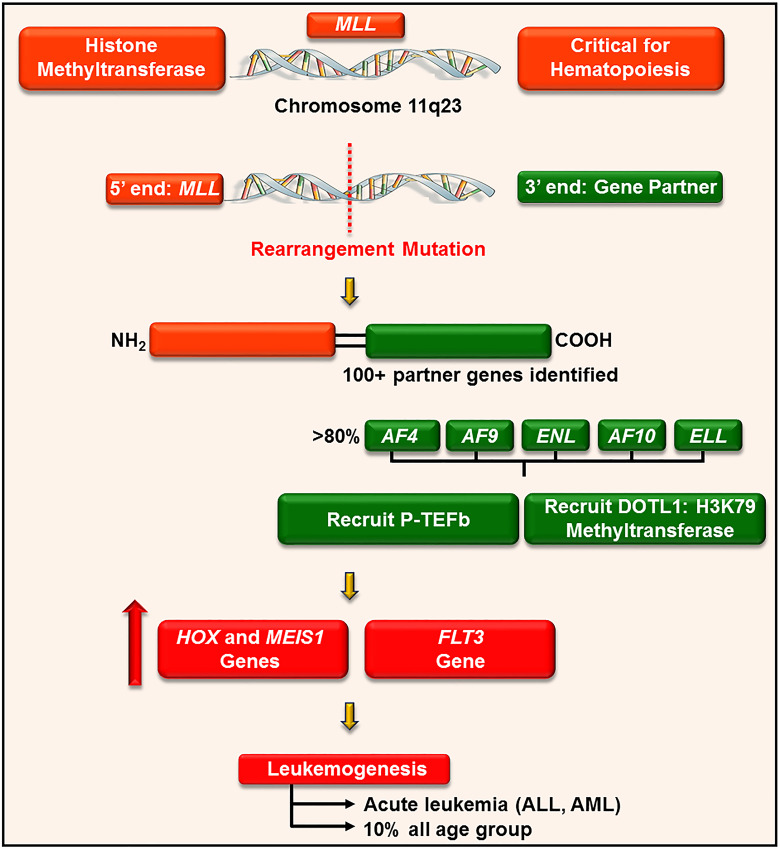
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous