

Hepatic Presentation of Late-Onset Multiple Acyl-CoA Dehydrogenase Deficiency (MADD): Case Report and Systematic Review
- PMID: 34041209
- PMCID: PMC8143529
- DOI: 10.3389/fped.2021.672004
Hepatic Presentation of Late-Onset Multiple Acyl-CoA Dehydrogenase Deficiency (MADD): Case Report and Systematic Review
Abstract
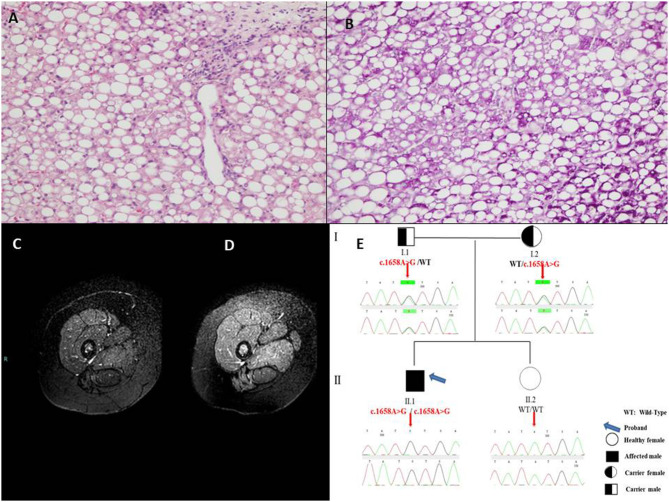
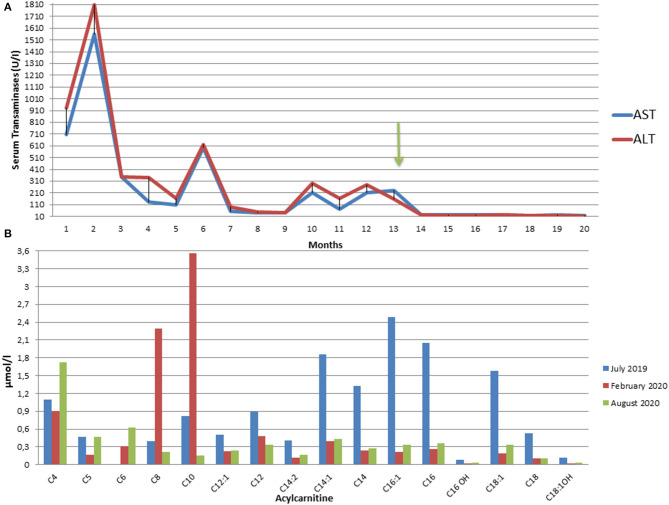
Diagnosis of pediatric steatohepatitis is a challenging issue due to a vast number of established and novel causes. Here, we report a child with Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) presenting with an underrated muscle weakness, exercise intolerance and an atypically severe steatotic liver involvement. A systematic literature review of liver involvement in MADD was performed as well. Our patient is a 11-year-old otherwise healthy, non-obese, male child admitted for some weakness/asthenia, vomiting and recurrent severe hypertransaminasemia (aspartate and alanine aminotransferases up to ×20 times upper limit of normal). Hepatic ultrasound showed a bright liver. MRI detected mild lipid storage of thighs muscles. A liver biopsy showed a micro-macrovacuolar steatohepatitis with minimal fibrosis. Main causes of hypertransaminasemia were ruled out. Serum aminoacids (increased proline), acylcarnitines (increased C4-C18) and a large excretion of urinary glutaric acid, ethylmalonic, butyric, isobutyric, 2-methyl-butyric and isovaleric acids suggested a diagnosis of MADD. Serum acylcarnitines and urinary organic acids fluctuated overtime paralleling serum transaminases during periods of illness/catabolic stress, confirming their recurrent nature. Genetic testing confirmed the diagnosis [homozygous c.1658A > G (p.Tyr553Cys) in exon 12 of the ETFDH gene]. Lipid-restricted diet and riboflavin treatment rapidly ameliorated symptoms, hepatic ultrasonography/enzymes, and metabolic profiles. Literature review (37 retrieved eligible studies, 283 patients) showed that liver is an extramuscular organ rarely involved in late-onset MADD (70 patients), and that amongst 45 patients who had fatty liver only nine had severe presentation. Conclusion: MADD is a disorder with a clinically heterogeneous phenotype. Our study suggests that MADD warrants consideration in the work-up of obesity-unrelated severe steatohepatitis.
Keywords: MADD; case report; fatty liver; hypertransaminasemia; steatohepatitis.
Copyright © 2021 Siano, Mandato, Nazzaro, Iannicelli, Ciccarelli, Barretta, Mazzaccara, Ruoppolo, Frisso, Baldi, Tartaglione, Di Salle, Melis and Vajro.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous