

New Interface for Faster Proteoform Analysis: Immunoprecipitation Coupled with SampleStream-Mass Spectrometry
- PMID: 34043341
- PMCID: PMC8530194
- DOI: 10.1021/jasms.1c00026
New Interface for Faster Proteoform Analysis: Immunoprecipitation Coupled with SampleStream-Mass Spectrometry
Abstract
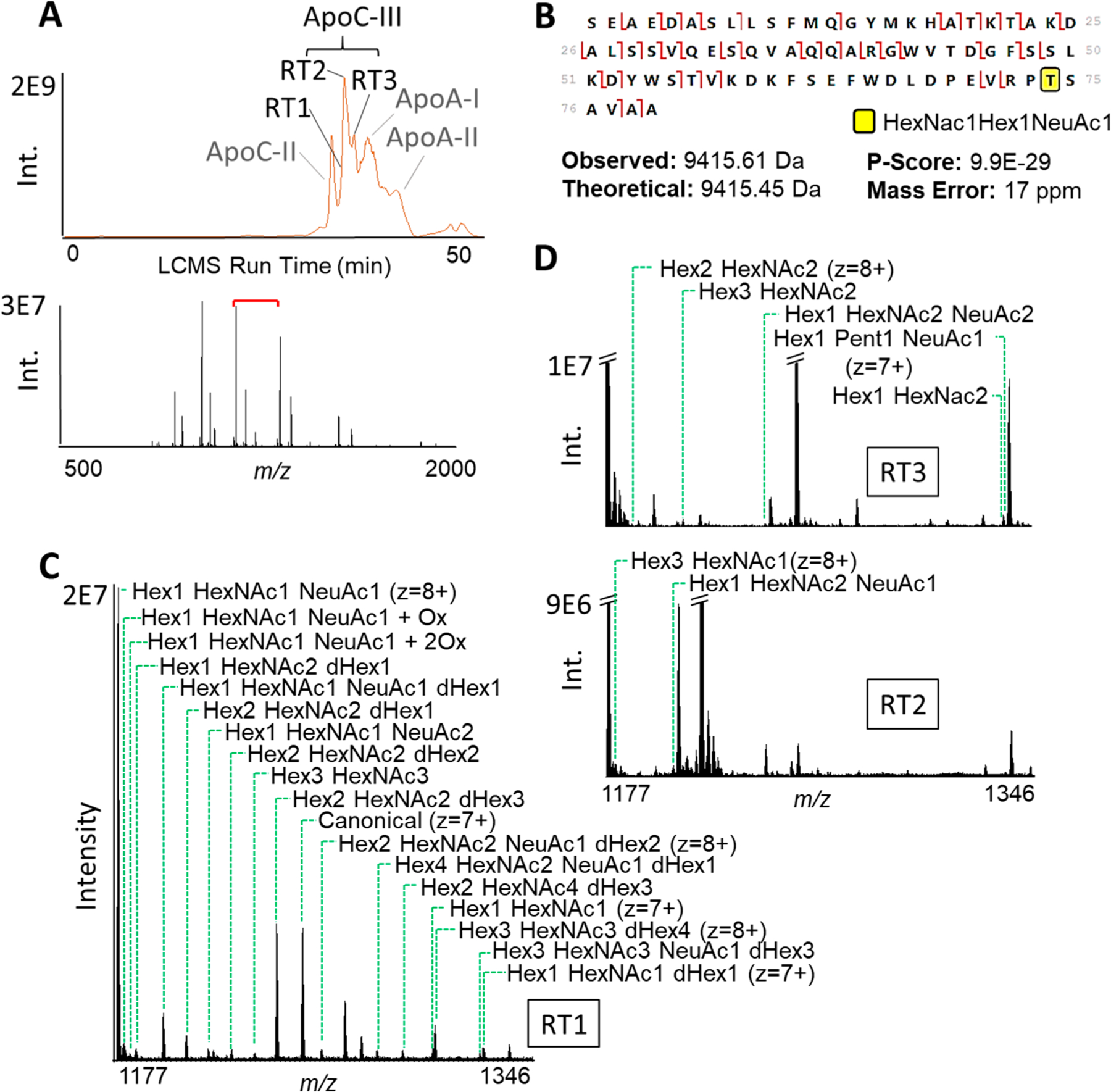
Different proteoform products of the same gene can exhibit differing associations with health and disease, and their patterns of modifications may offer more precise markers of phenotypic differences between individuals. However, currently employed protein-biomarker discovery and quantification tools, such as bottom-up proteomics and ELISAs, are mostly proteoform-unaware. Moreover, the current throughput for proteoform-level analyses by liquid chromatography mass spectrometry (LCMS) for quantitative top-down proteomics is incompatible with population-level biomarker surveys requiring robust, faster proteoform analysis. To this end, we developed immunoprecipitation coupled to SampleStream mass spectrometry (IP-SampleStream-MS) as a high-throughput, automated technique for the targeted quantification of proteoforms. We applied IP-SampleStream-MS to serum samples of 25 individuals to assess the proteoform abundances of apolipoproteins A-I (ApoA-I) and C-III (ApoC-III). The results for ApoA-I were compared to those of LCMS for these individuals, with IP-SampleStream-MS showing a >7-fold higher throughput with >50% better analytical variation. Proteoform abundances measured by IP-SampleStream-MS correlated strongly to LCMS-based values (R2 = 0.6-0.9) and produced convergent proteoform-to-phenotype associations, namely, the abundance of canonical ApoA-I was associated with lower HDL-C (R = 0.5) and glycated ApoA-I with higher fasting glucose (R = 0.6). We also observed proteoform-to-phenotype associations for ApoC-III, 22 glycoproteoforms of which were characterized in this study. The abundance of ApoC-III modified by a single N-acetyl hexosamine (HexNAc) was associated with indices of obesity, such as BMI, weight, and waist circumference (R ∼ 0.7). These data show IP-SampleStream-MS to be a robust, scalable workflow for high-throughput associations of proteoforms to phenotypes.
Conflict of interest statement
The authors declare the following competing financial interest(s): Authors N.L.K. and P.D.C. report a conflict of interest with Integrated Protein Technologies, the supplier of the SampleStream system used in this work.
Figures





References
-
- Seckler HDS; Fornelli L; Mutharasan RK; Thaxton CS; Fellers R; Daviglus M; Sniderman A; Rader D; Kelleher NL; Lloyd-Jones DM; Compton PD; Wilkins JT A Targeted, Differential Top-Down Proteomic Methodology for Comparison of ApoA-I Proteoforms in Individuals with High and Low HDL Efflux Capacity. J. Proteome Res 2018, 17 (6), 2156–2164. - PMC - PubMed
-
- Tucholski T; Cai W; Gregorich ZR; Bayne EF; Mitchell SD; McIlwain SJ; de Lange WJ; Wrobbel M; Karp H; Hite Z; Vikhorev PG; Marston SB; Lal S; Li A; Dos Remedios C; Kohmoto T; Hermsen J; Ralphe JC; Kamp TJ; Moss RL; Ge Y Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (40), 24691–24700. - PMC - PubMed
-
- Ntai I; LeDuc RD; Fellers RT; Erdmann-Gilmore P; Davies SR; Rumsey J; Early BP; Thomas PM; Li S; Compton PD; Ellis MJ; Ruggles KV; Fenyo D; Boja ES; Rodriguez H; Townsend RR; Kelleher NL Integrated Bottom-Up and Top-Down Proteomics of Patient-Derived Breast Tumor Xenografts. Mol. Cell Proteomics 2016, 15 (1), 45–56. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
