

Inferring Genome-Wide Correlations of Mutation Fitness Effects between Populations
- PMID: 34043790
- PMCID: PMC8476148
- DOI: 10.1093/molbev/msab162
Inferring Genome-Wide Correlations of Mutation Fitness Effects between Populations
Abstract
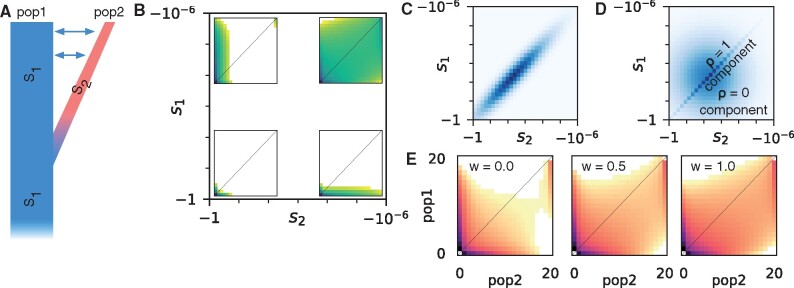
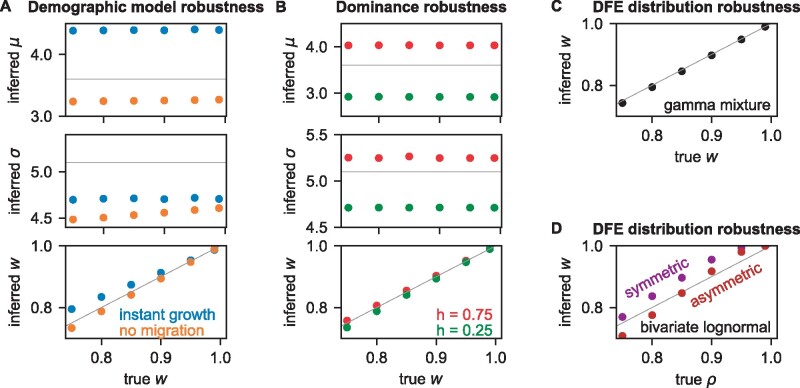
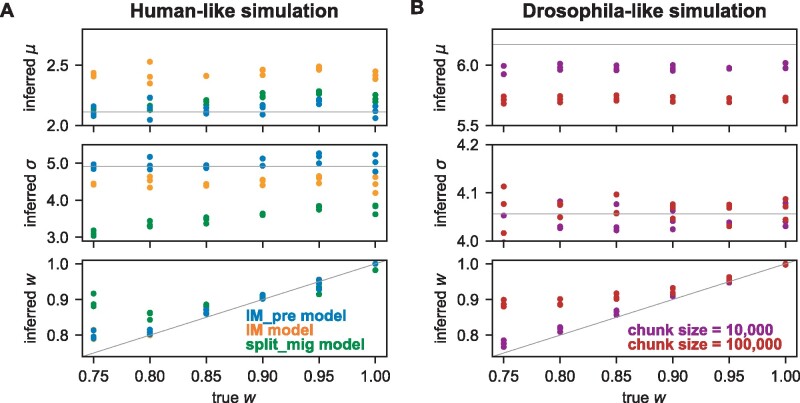
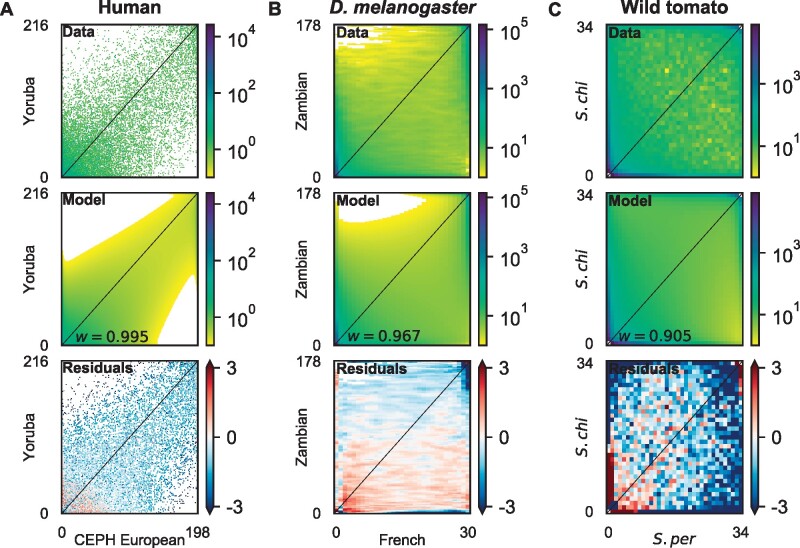
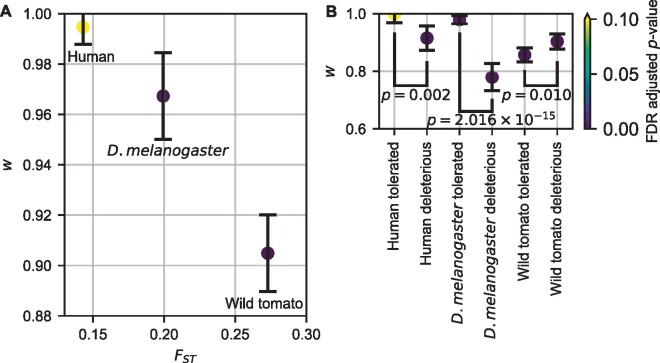
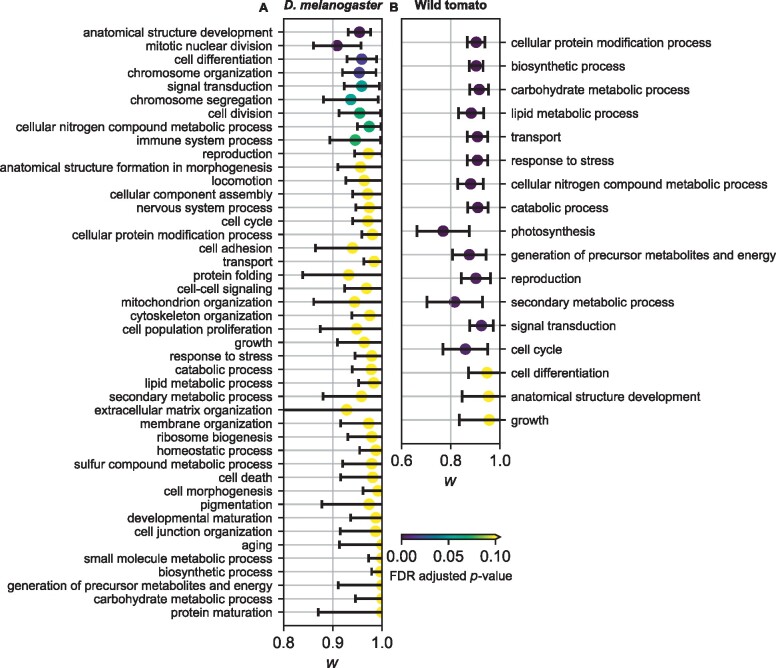
The effect of a mutation on fitness may differ between populations depending on environmental and genetic context, but little is known about the factors that underlie such differences. To quantify genome-wide correlations in mutation fitness effects, we developed a novel concept called a joint distribution of fitness effects (DFE) between populations. We then proposed a new statistic w to measure the DFE correlation between populations. Using simulation, we showed that inferring the DFE correlation from the joint allele frequency spectrum is statistically precise and robust. Using population genomic data, we inferred DFE correlations of populations in humans, Drosophila melanogaster, and wild tomatoes. In these species, we found that the overall correlation of the joint DFE was inversely related to genetic differentiation. In humans and D. melanogaster, deleterious mutations had a lower DFE correlation than tolerated mutations, indicating a complex joint DFE. Altogether, the DFE correlation can be reliably inferred, and it offers extensive insight into the genetics of population divergence.
Keywords: distribution of fitness effects; population divergence; population genetics.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
