

Molecular Mechanisms of Renal Magnesium Reabsorption
- PMID: 34045316
- PMCID: PMC8729834
- DOI: 10.1681/ASN.2021010042
Molecular Mechanisms of Renal Magnesium Reabsorption
Abstract
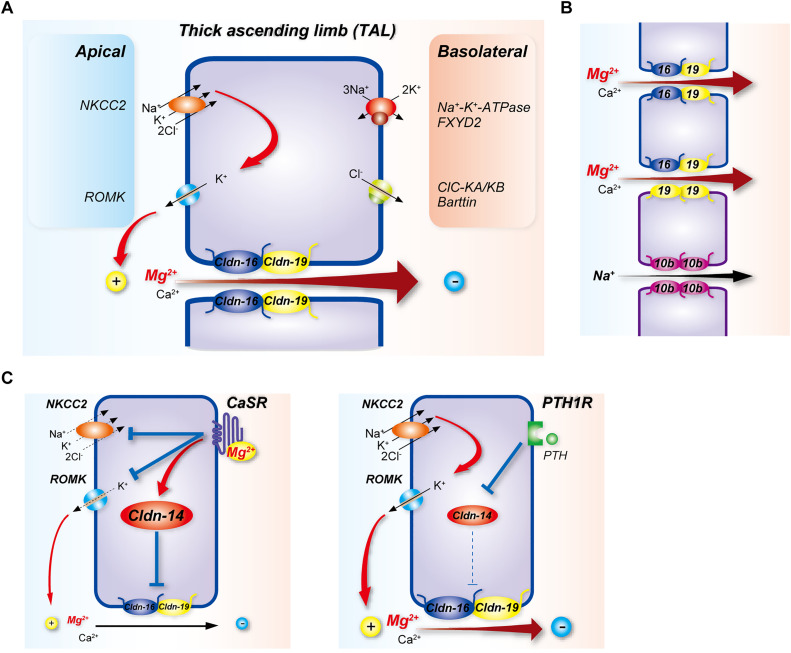
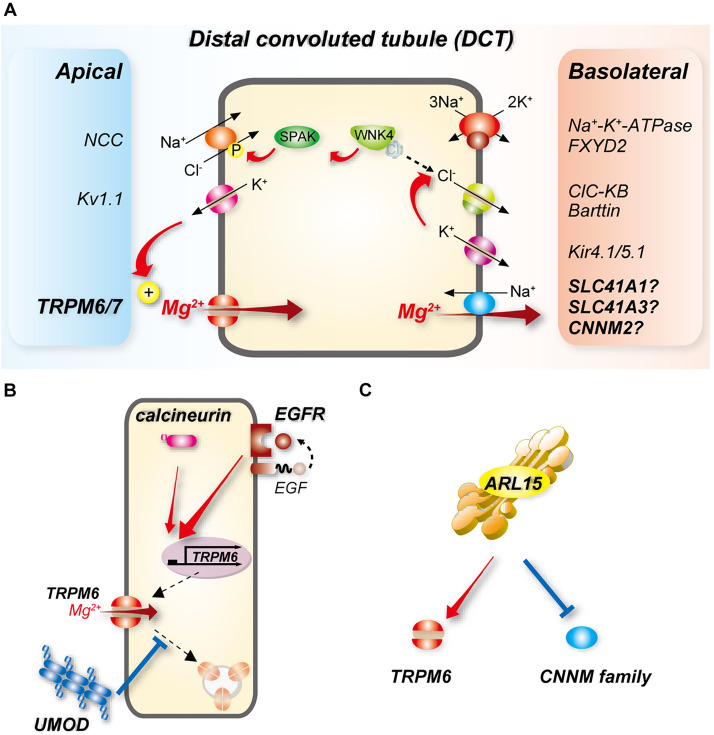
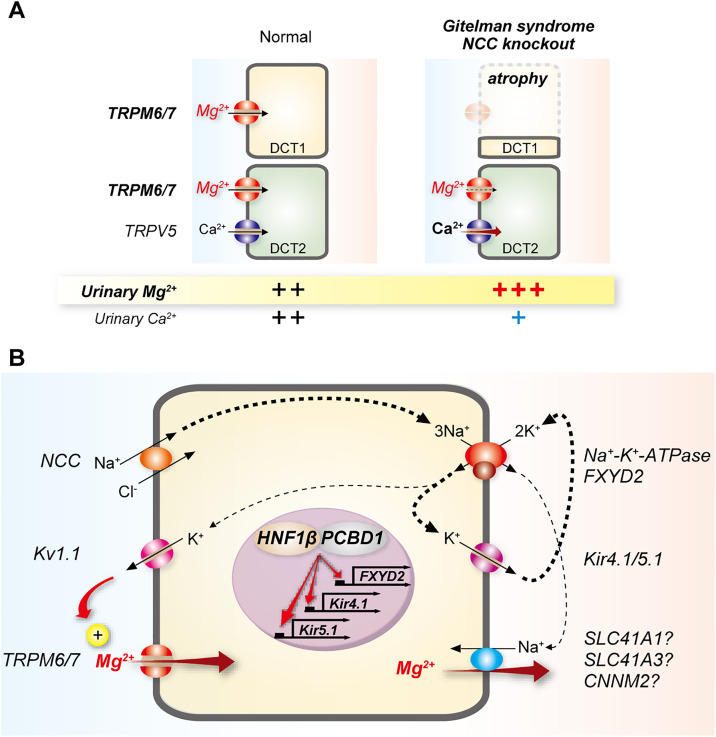
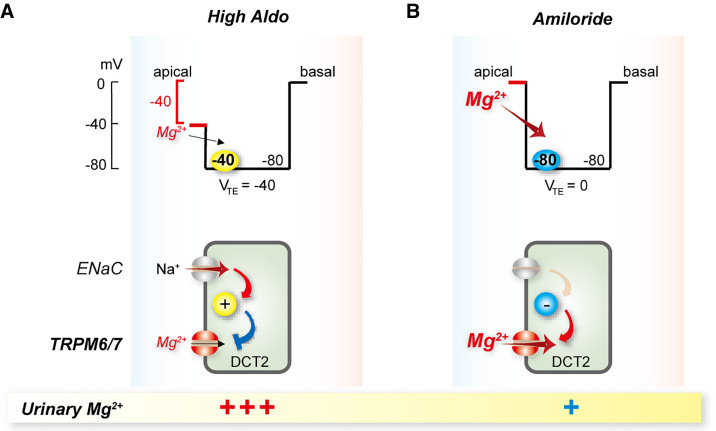
Magnesium is an essential cofactor in many cellular processes, and aberrations in magnesium homeostasis can have life-threatening consequences. The kidney plays a central role in maintaining serum magnesium within a narrow range (0.70-1.10 mmol/L). Along the proximal tubule and thick ascending limb, magnesium reabsorption occurs via paracellular pathways. Members of the claudin family form the magnesium pores in these segments, and also regulate magnesium reabsorption by adjusting the transepithelial voltage that drives it. Along the distal convoluted tubule transcellular reabsorption via heteromeric TRPM6/7 channels predominates, although paracellular reabsorption may also occur. In this segment, the NaCl cotransporter plays a critical role in determining transcellular magnesium reabsorption. Although the general machinery involved in renal magnesium reabsorption has been identified by studying genetic forms of magnesium imbalance, the mechanisms regulating it are poorly understood. This review discusses pathways of renal magnesium reabsorption by different segments of the nephron, emphasizing newer findings that provide insight into regulatory process, and outlining critical unanswered questions.
Keywords: Gitelman syndrome; cell and transport physiology; claudin; ion transport; magnesium.
Copyright © 2021 by the American Society of Nephrology.
Figures
References
-
- de Baaij JH, Hoenderop JG, Bindels RJ: Magnesium in man: implications for health and disease. Physiol Rev 95: 1–46, 2015 - PubMed
-
- Wong NL, Dirks JH, Quamme GA: Tubular reabsorptive capacity for magnesium in the dog kidney. Am J Physiol 244: F78–F83, 1983 - PubMed
-
- Wong NL, Whiting SJ, Mizgala CL, Quamme GA: Electrolyte handling by the superficial nephron of the rabbit. Am J Physiol 250: F590–F595, 1986 - PubMed
-
- Brunette MG, Vigneault N, Carriere S: Micropuncture study of magnesium transport along the nephron in the young rat. Am J Physiol 227: 891–896, 1974 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
