

Mitochondrial and metabolic dysfunction in Friedreich ataxia: update on pathophysiological relevance and clinical interventions
- PMID: 34046211
- PMCID: PMC8132591
- DOI: 10.1042/NS20200093
Mitochondrial and metabolic dysfunction in Friedreich ataxia: update on pathophysiological relevance and clinical interventions
Abstract
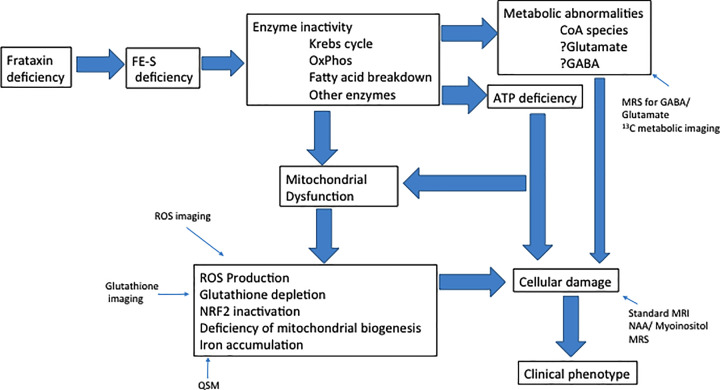
Friedreich ataxia (FRDA) is a recessive disorder resulting from relative deficiency of the mitochondrial protein frataxin. Frataxin functions in the process of iron-sulfur (Fe-S) cluster synthesis. In this review, we update some of the processes downstream of frataxin deficiency that may mediate the pathophysiology. Based on cellular models, in vivo models and observations of patients, ferroptosis may play a major role in the pathogenesis of FRDA along with depletion of antioxidant reserves and abnormalities of mitochondrial biogenesis. Ongoing clinical trials with ferroptosis inhibitors and nuclear factor erythroid 2-related factor 2 (Nrf2) activators are now targeting each of the processes. In addition, better understanding of the mitochondrial events in FRDA may allow the development of improved imaging methodology for assessing the disorder. Though not technologically feasible at present, metabolic imaging approaches may provide a direct methodology to understand the mitochondrial changes occurring in FRDA and provide a methodology to monitor upcoming trials of frataxin restoration.
Keywords: biomarkers; clinical trial; mitochondrial biogenesis.
© 2021 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous