

The Role of RNA Methyltransferase METTL3 in Hepatocellular Carcinoma: Results and Perspectives
- PMID: 34046411
- PMCID: PMC8144501
- DOI: 10.3389/fcell.2021.674919
The Role of RNA Methyltransferase METTL3 in Hepatocellular Carcinoma: Results and Perspectives
Abstract
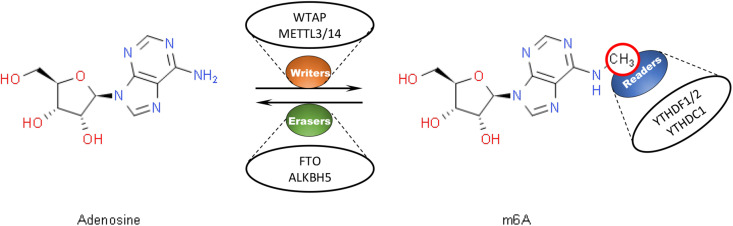
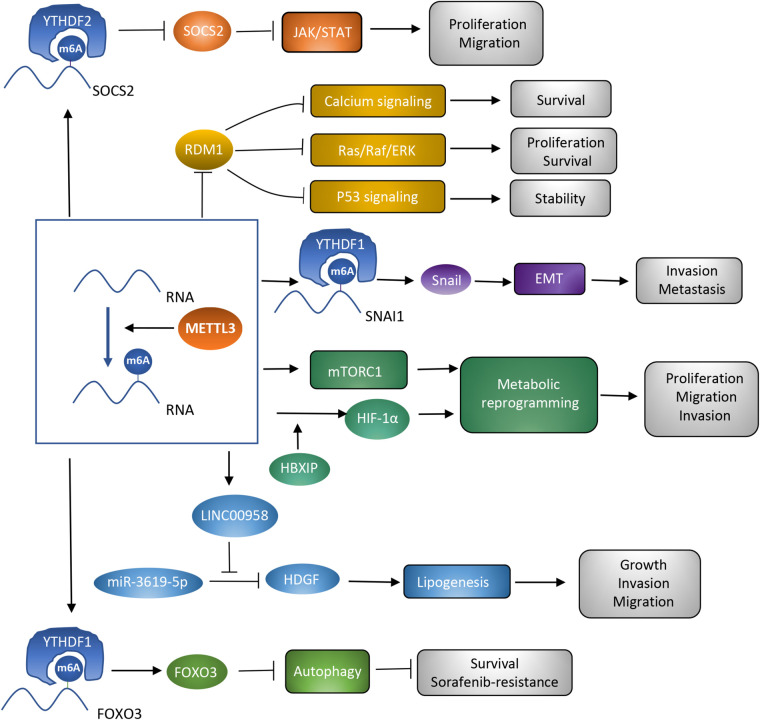
Hepatocellular carcinoma (HCC) is the 6th most prevalent cancer and the 4th leading cause of cancer-related death worldwide. Mechanisms explaining the carcinogenesis of HCC are not clear yet. In recent years, rapid development of N6-methyladenosine (m6A) modification provides a fresh approach to disclosing this mystery. As the most prevalent mRNA modification in eukaryotes, m6A modification is capable to post-transcriptionally affect RNA splicing, stability, and translation, thus participating in a variety of biological and pathological processes including cell proliferation, apoptosis, tumor invasion and metastasis. METTL3 has been recognized as a pivotal methyltransferase and essential to the performance of m6A modification. METTL3 can regulate RNA expression in a m6A-dependent manner and contribute to the carcinogenesis, tumor progression, and drug resistance of HCC. In the present review, we are going to make a clear summary of the known roles of METTL3 in HCC, and explicitly narrate the potential mechanisms for these roles.
Keywords: Hepatocellular carcinoma; METTL3; RNA modification; drug-resistance; m6A.
Copyright © 2021 Pan, Lin, Hao, Chu, Wan and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Bokar J. A. (2005). “The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA,” in Fine-Tuning of RNA Functions by Modification and Editing, ed. Grosjean H. (Berlin: Springer Berlin Heidelberg; ), 141–177.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials