

A map of the SARS-CoV-2 RNA structurome
- PMID: 34046592
- PMCID: PMC8140738
- DOI: 10.1093/nargab/lqab043
A map of the SARS-CoV-2 RNA structurome
Abstract
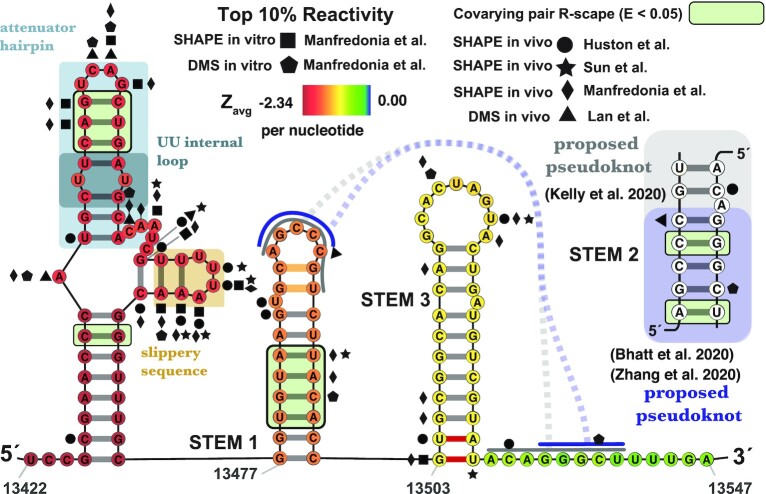
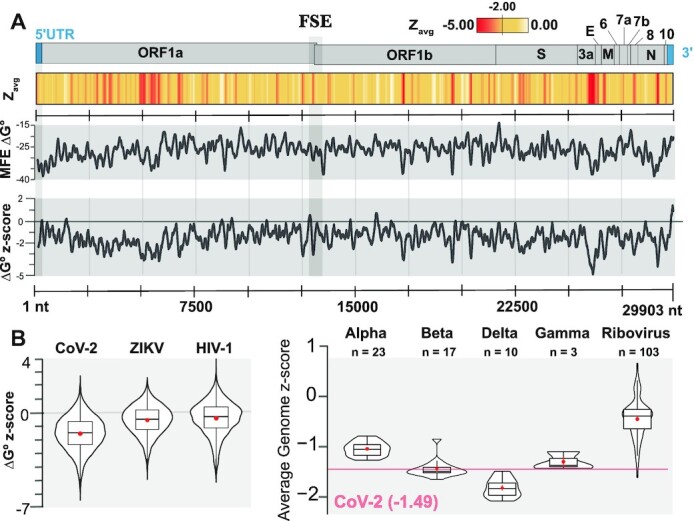
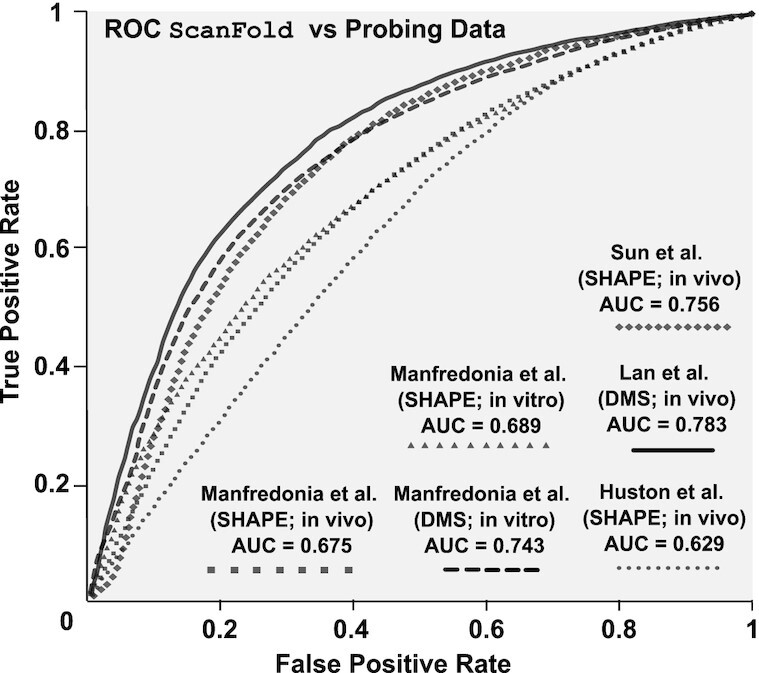
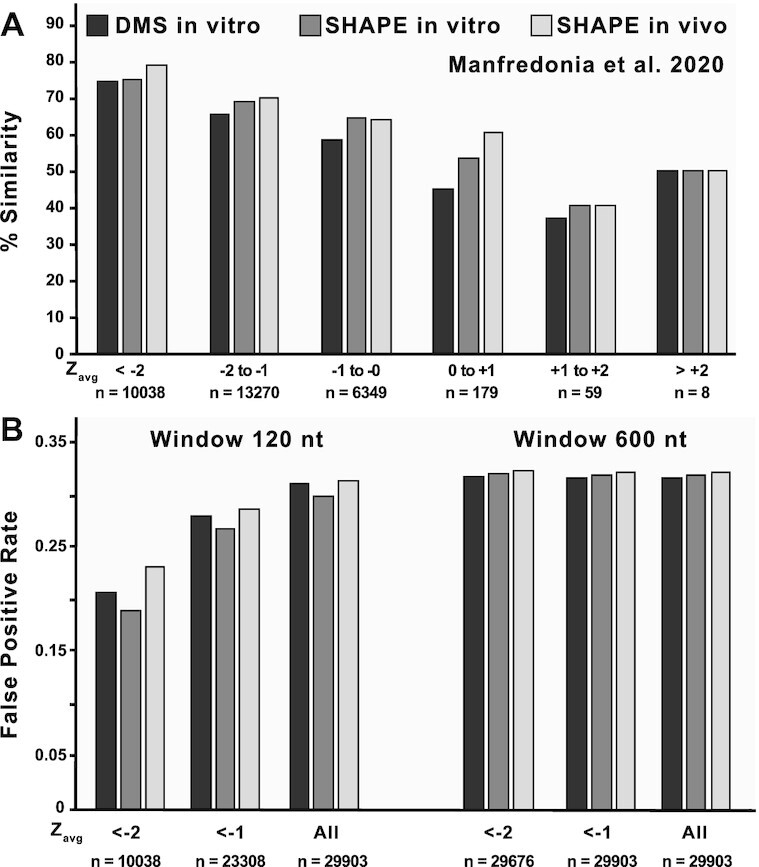
SARS-CoV-2 has exploded throughout the human population. To facilitate efforts to gain insights into SARS-CoV-2 biology and to target the virus therapeutically, it is essential to have a roadmap of likely functional regions embedded in its RNA genome. In this report, we used a bioinformatics approach, ScanFold, to deduce the local RNA structural landscape of the SARS-CoV-2 genome with the highest likelihood of being functional. We recapitulate previously-known elements of RNA structure and provide a model for the folding of an essential frameshift signal. Our results find that SARS-CoV-2 is greatly enriched in unusually stable and likely evolutionarily ordered RNA structure, which provides a large reservoir of potential drug targets for RNA-binding small molecules. Results are enhanced via the re-analyses of publicly-available genome-wide biochemical structure probing datasets that are broadly in agreement with our models. Additionally, ScanFold was updated to incorporate experimental data as constraints in the analysis to facilitate comparisons between ScanFold and other RNA modelling approaches. Ultimately, ScanFold was able to identify eight highly structured/conserved motifs in SARS-CoV-2 that agree with experimental data, without explicitly using these data. All results are made available via a public database (the RNAStructuromeDB: https://structurome.bb.iastate.edu/sars-cov-2) and model comparisons are readily viewable at https://structurome.bb.iastate.edu/sars-cov-2-global-model-comparisons.
© The Author(s) 2021. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
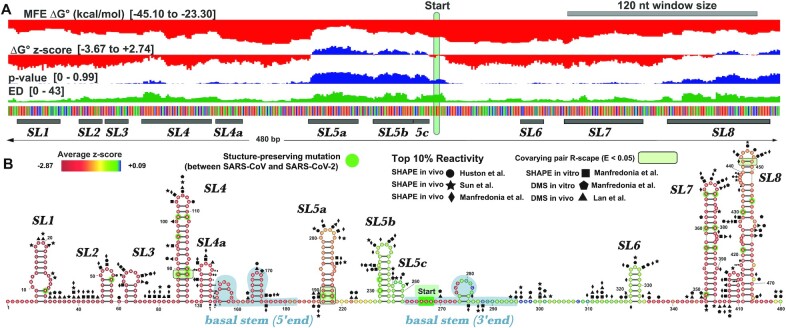
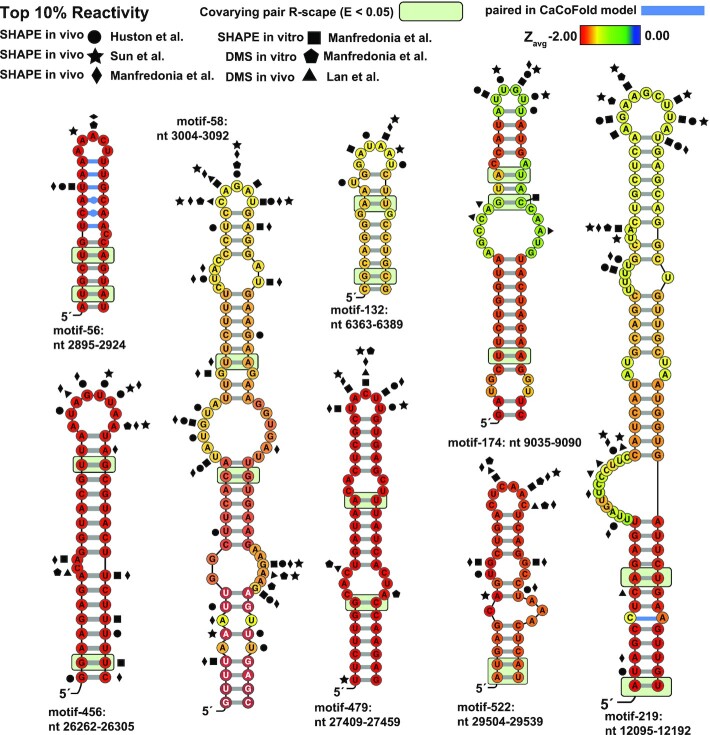
Update of
-
An in silico map of the SARS-CoV-2 RNA Structurome.bioRxiv [Preprint]. 2020 Apr 18:2020.04.17.045161. doi: 10.1101/2020.04.17.045161. bioRxiv. 2020. Update in: NAR Genom Bioinform. 2021 May 22;3(2):lqab043. doi: 10.1093/nargab/lqab043. PMID: 32511381 Free PMC article. Updated. Preprint.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
