

Assessing the functional relevance of splice isoforms
- PMID: 34046593
- PMCID: PMC8140736
- DOI: 10.1093/nargab/lqab044
Assessing the functional relevance of splice isoforms
Abstract
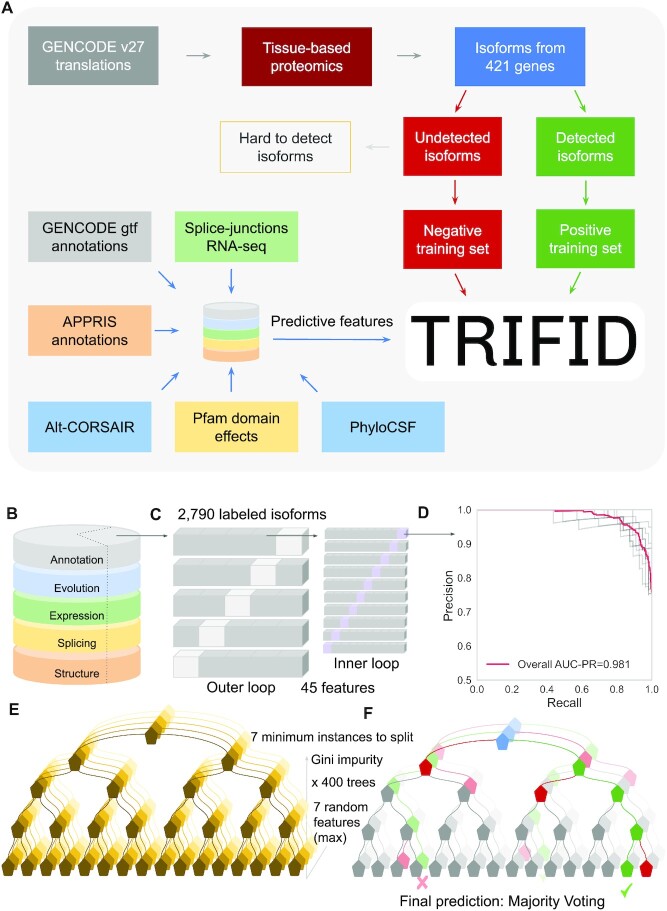
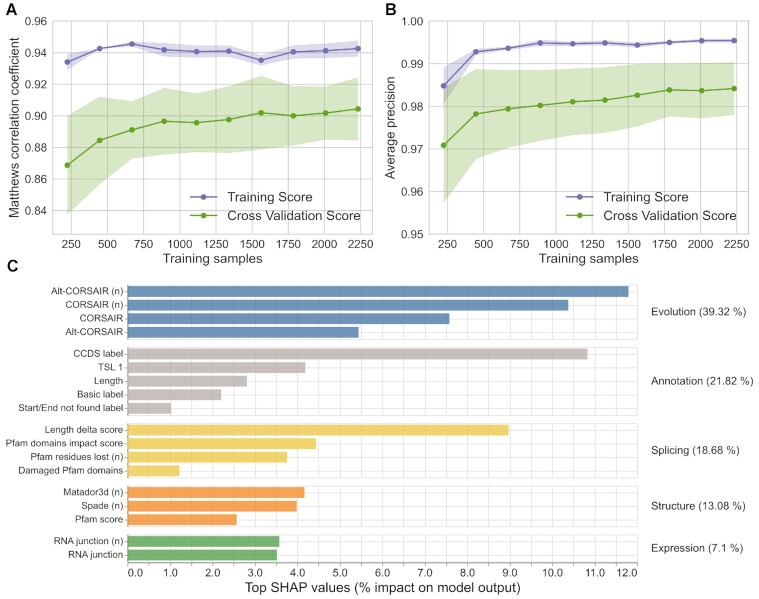
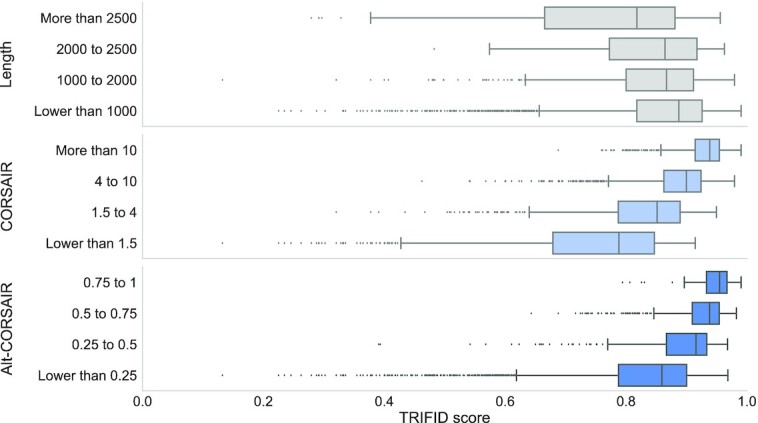
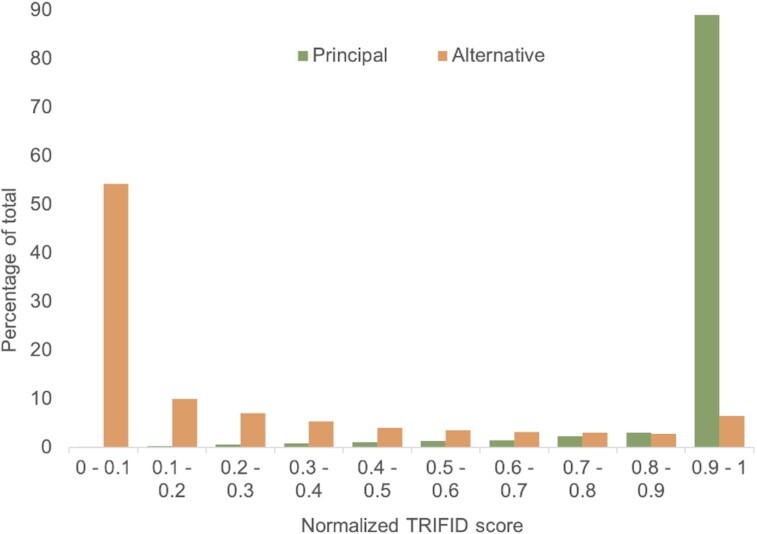
Alternative splicing of messenger RNA can generate an array of mature transcripts, but it is not clear how many go on to produce functionally relevant protein isoforms. There is only limited evidence for alternative proteins in proteomics analyses and data from population genetic variation studies indicate that most alternative exons are evolving neutrally. Determining which transcripts produce biologically important isoforms is key to understanding isoform function and to interpreting the real impact of somatic mutations and germline variations. Here we have developed a method, TRIFID, to classify the functional importance of splice isoforms. TRIFID was trained on isoforms detected in large-scale proteomics analyses and distinguishes these biologically important splice isoforms with high confidence. Isoforms predicted as functionally important by the algorithm had measurable cross species conservation and significantly fewer broken functional domains. Additionally, exons that code for these functionally important protein isoforms are under purifying selection, while exons from low scoring transcripts largely appear to be evolving neutrally. TRIFID has been developed for the human genome, but it could in principle be applied to other well-annotated species. We believe that this method will generate valuable insights into the cellular importance of alternative splicing.
© The Author(s) 2021. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
Similar articles
-
Alternatively Spliced Homologous Exons Have Ancient Origins and Are Highly Expressed at the Protein Level.PLoS Comput Biol. 2015 Jun 10;11(6):e1004325. doi: 10.1371/journal.pcbi.1004325. eCollection 2015 Jun. PLoS Comput Biol. 2015. PMID: 26061177 Free PMC article.
-
Alternative Splicing May Not Be the Key to Proteome Complexity.Trends Biochem Sci. 2017 Feb;42(2):98-110. doi: 10.1016/j.tibs.2016.08.008. Epub 2016 Oct 3. Trends Biochem Sci. 2017. PMID: 27712956 Free PMC article. Review.
-
APPRIS principal isoforms and MANE Select transcripts define reference splice variants.Bioinformatics. 2022 Sep 16;38(Suppl_2):ii89-ii94. doi: 10.1093/bioinformatics/btac473. Bioinformatics. 2022. PMID: 36124785 Free PMC article.
-
APPRIS: selecting functionally important isoforms.Nucleic Acids Res. 2022 Jan 7;50(D1):D54-D59. doi: 10.1093/nar/gkab1058. Nucleic Acids Res. 2022. PMID: 34755885 Free PMC article.
-
Cancer-Associated Perturbations in Alternative Pre-messenger RNA Splicing.Cancer Treat Res. 2013;158:41-94. doi: 10.1007/978-3-642-31659-3_3. Cancer Treat Res. 2013. PMID: 24222354 Review.
Cited by
-
Identification of senescent cell subpopulations by CITE-seq analysis.Aging Cell. 2024 Nov;23(11):e14297. doi: 10.1111/acel.14297. Epub 2024 Aug 14. Aging Cell. 2024. PMID: 39143693 Free PMC article.
-
Transcriptomic Profiling Provides Insight into the Molecular Basis of Heterosis in Philippine-Reared Bombyx mori Hybrids.Insects. 2025 Feb 26;16(3):243. doi: 10.3390/insects16030243. Insects. 2025. PMID: 40266772 Free PMC article.
-
Ensembl 2024.Nucleic Acids Res. 2024 Jan 5;52(D1):D891-D899. doi: 10.1093/nar/gkad1049. Nucleic Acids Res. 2024. PMID: 37953337 Free PMC article.
-
Toward a comprehensive profiling of alternative splicing proteoform structures, interactions and functions.Curr Opin Struct Biol. 2025 Feb;90:102979. doi: 10.1016/j.sbi.2024.102979. Epub 2025 Jan 7. Curr Opin Struct Biol. 2025. PMID: 39778413 Free PMC article. Review.
-
Profiling genetically driven alternative splicing across the Indonesian archipelago.Am J Hum Genet. 2024 Nov 7;111(11):2458-2477. doi: 10.1016/j.ajhg.2024.09.004. Epub 2024 Oct 8. Am J Hum Genet. 2024. PMID: 39383868 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
