

Recent progress in mass spectrometry-based strategies for elucidating protein-protein interactions
- PMID: 34046695
- PMCID: PMC8159249
- DOI: 10.1007/s00018-021-03856-0
Recent progress in mass spectrometry-based strategies for elucidating protein-protein interactions
Abstract
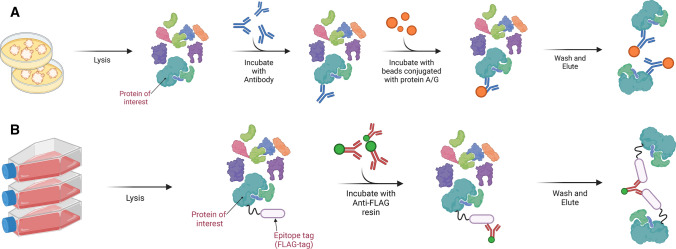
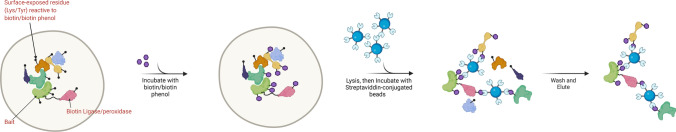
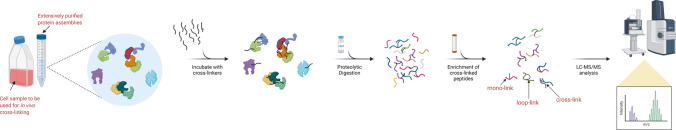
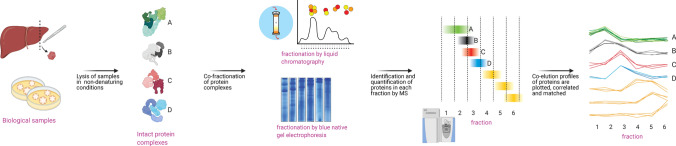
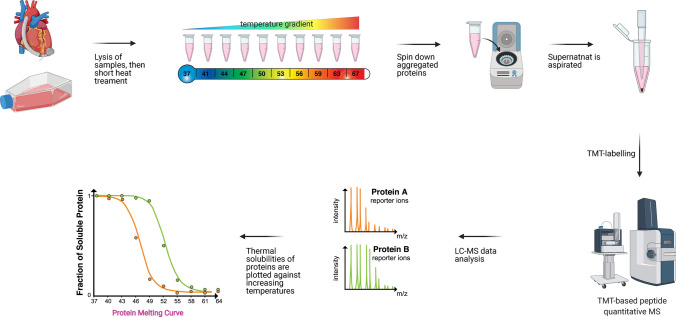
Protein-protein interactions are fundamental to various aspects of cell biology with many protein complexes participating in numerous fundamental biological processes such as transcription, translation and cell cycle. MS-based proteomics techniques are routinely applied for characterising the interactome, such as affinity purification coupled to mass spectrometry that has been used to selectively enrich and identify interacting partners of a bait protein. In recent years, many orthogonal MS-based techniques and approaches have surfaced including proximity-dependent labelling of neighbouring proteins, chemical cross-linking of two interacting proteins, as well as inferring PPIs from the co-behaviour of proteins such as the co-fractionating profiles and the thermal solubility profiles of proteins. This review discusses the underlying principles, advantages, limitations and experimental considerations of these emerging techniques. In addition, a brief account on how MS-based techniques are used to investigate the structural and functional properties of protein complexes, including their topology, stoichiometry, copy number and dynamics, are discussed.
Keywords: Affinity purification coupled to mass spectrometry (AP-MS); Co-fractionation mass spectrometry (coFrac-MS); Cross-linking mass spectrometry (XL-MS); Proximity-dependent biotinylation coupled to MS (PDB-MS); Thermal proximity coaggregation (TPCA).
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
-
- Wu Z, Malty R, Moutaoufik MT, et al. Advances in experimental medicine and biology. New York LLC: Springer; 2019. A tag-based affinity purification mass spectrometry workflow for systematic isolation of the human mitochondrial protein complexes; pp. 83–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
