

Inflammation drives alternative first exon usage to regulate immune genes including a novel iron-regulated isoform of Aim2
- PMID: 34047695
- PMCID: PMC8260223
- DOI: 10.7554/eLife.69431
Inflammation drives alternative first exon usage to regulate immune genes including a novel iron-regulated isoform of Aim2
Abstract
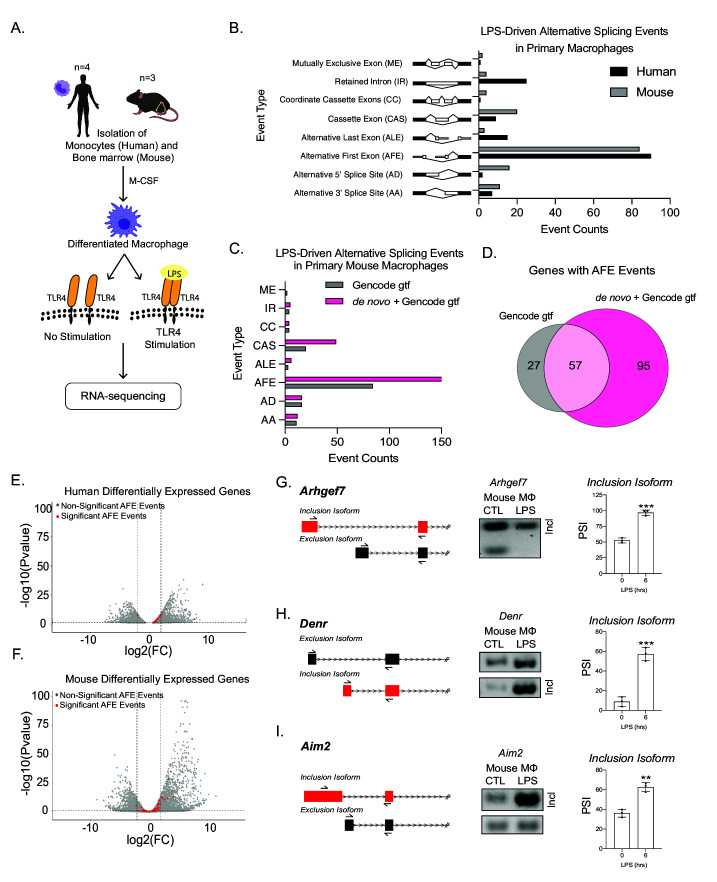
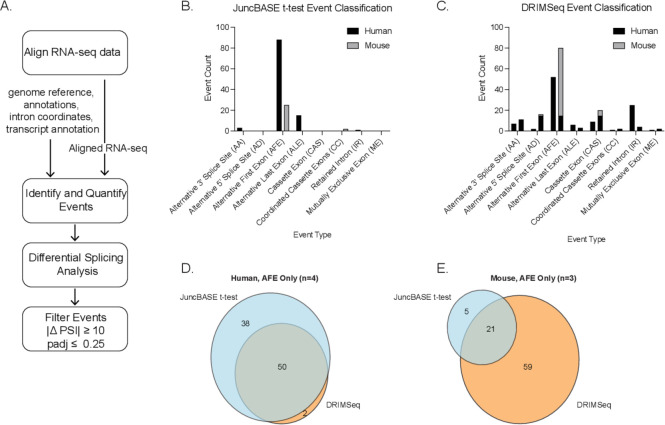
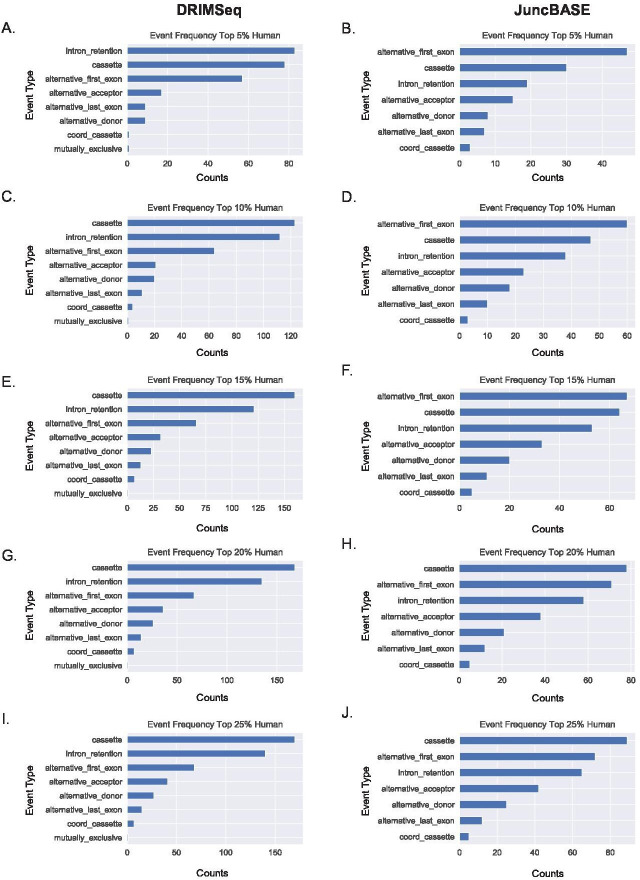
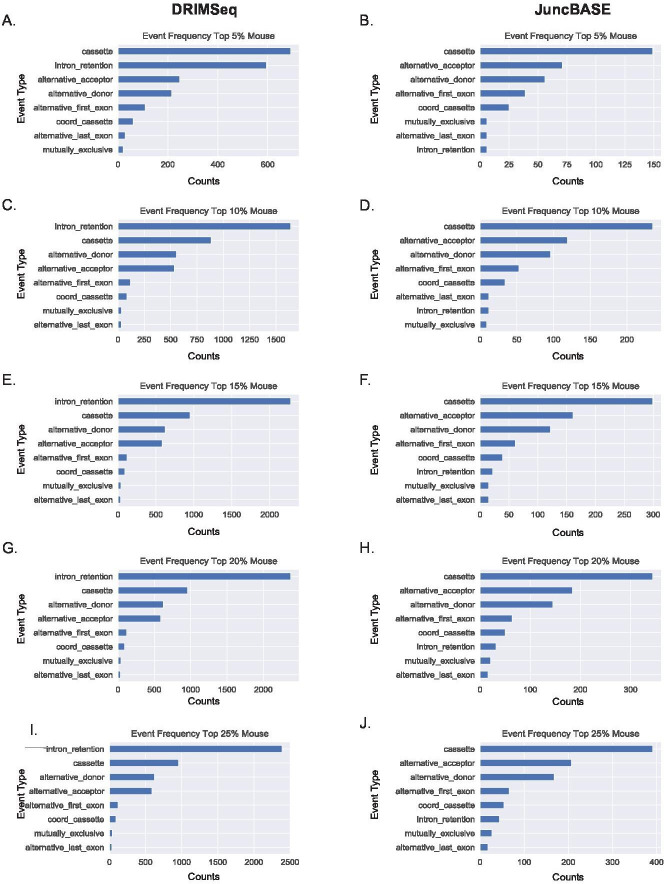
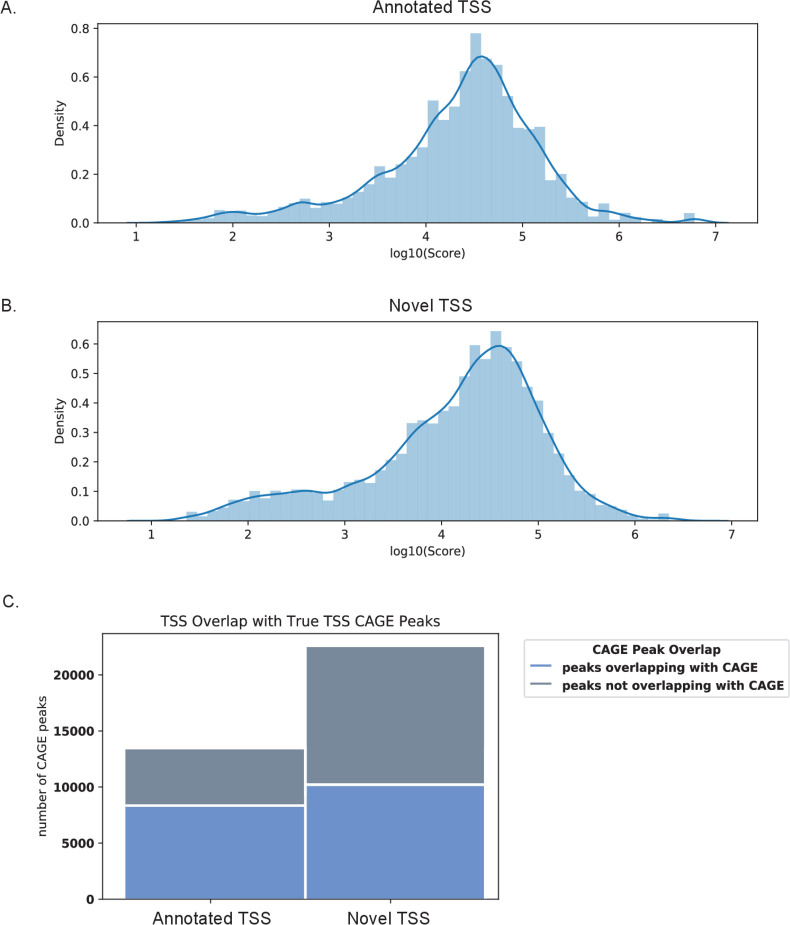
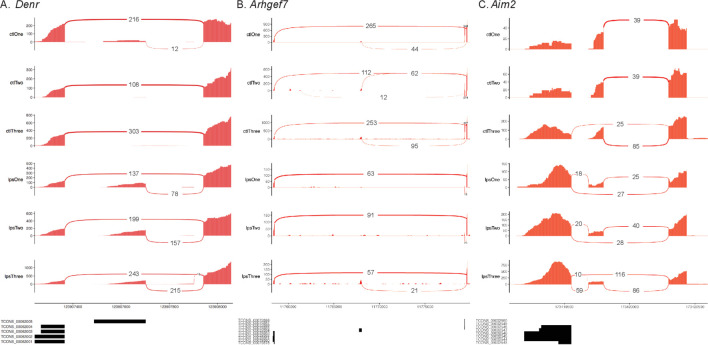
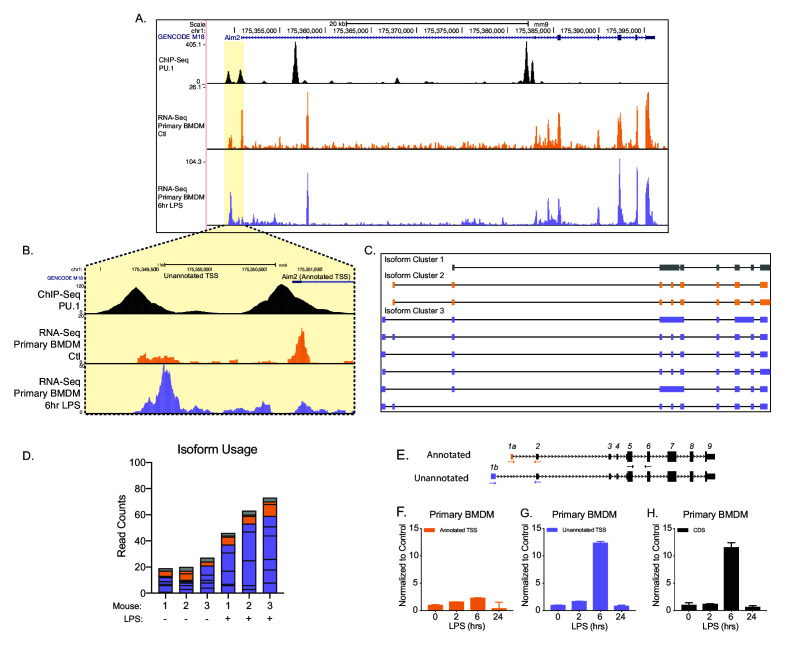
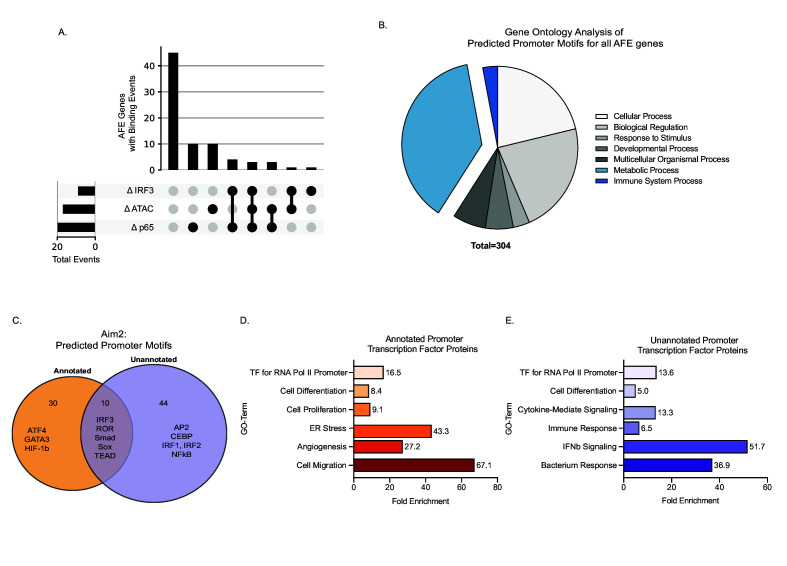
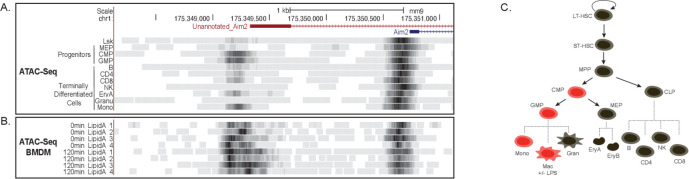
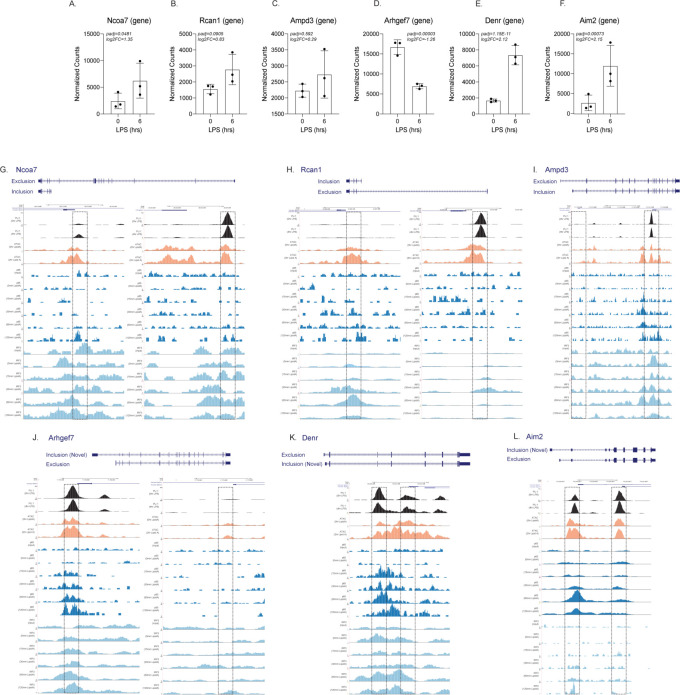
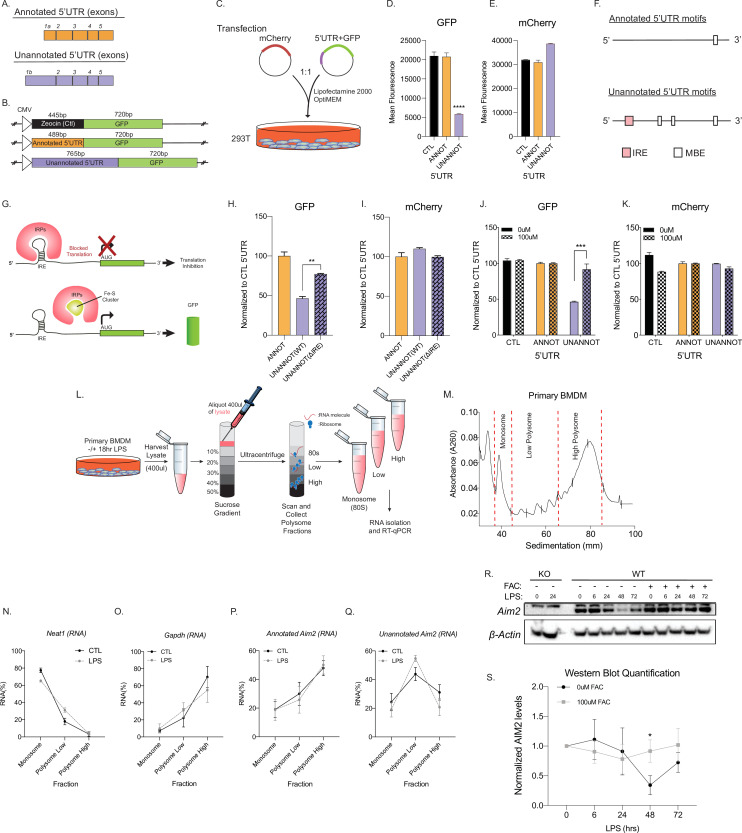
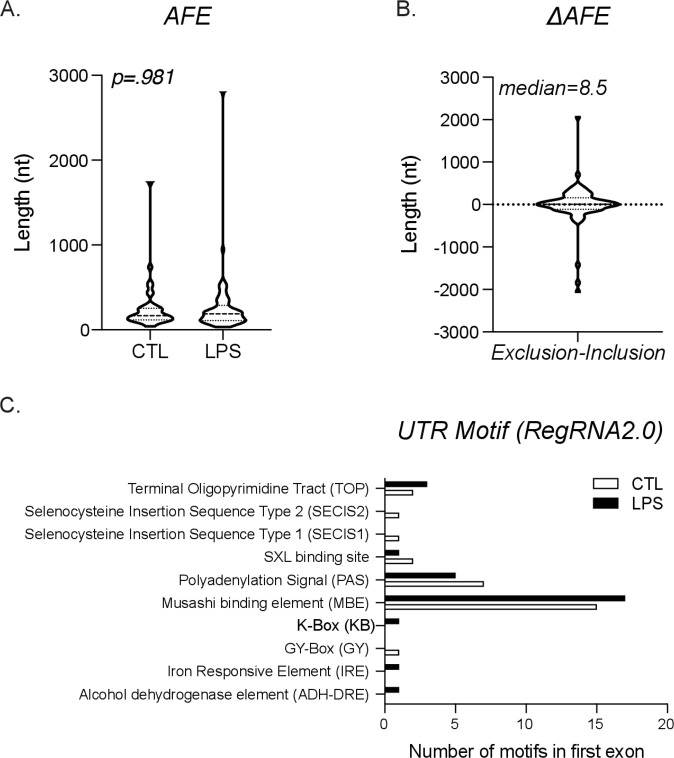
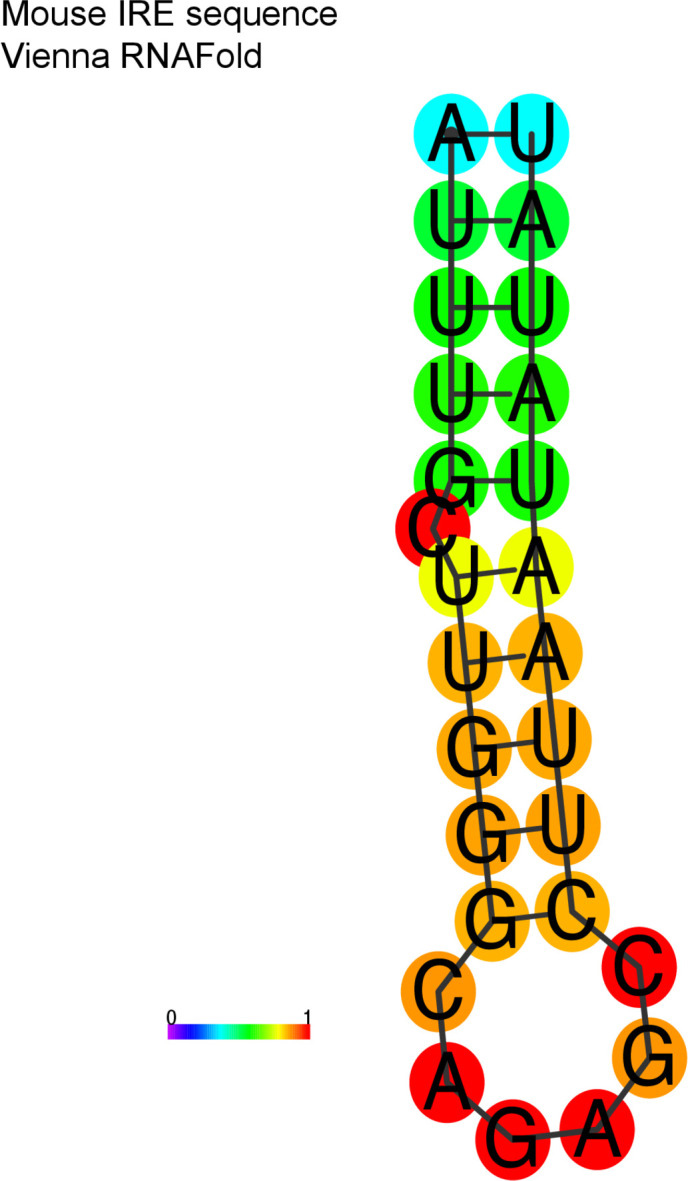
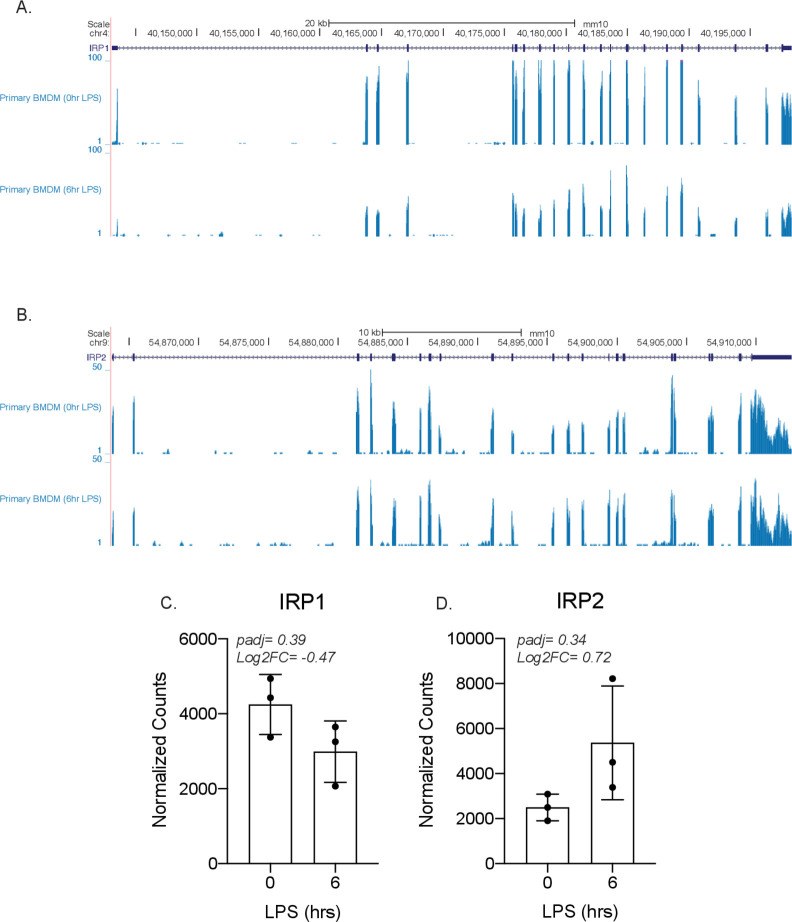
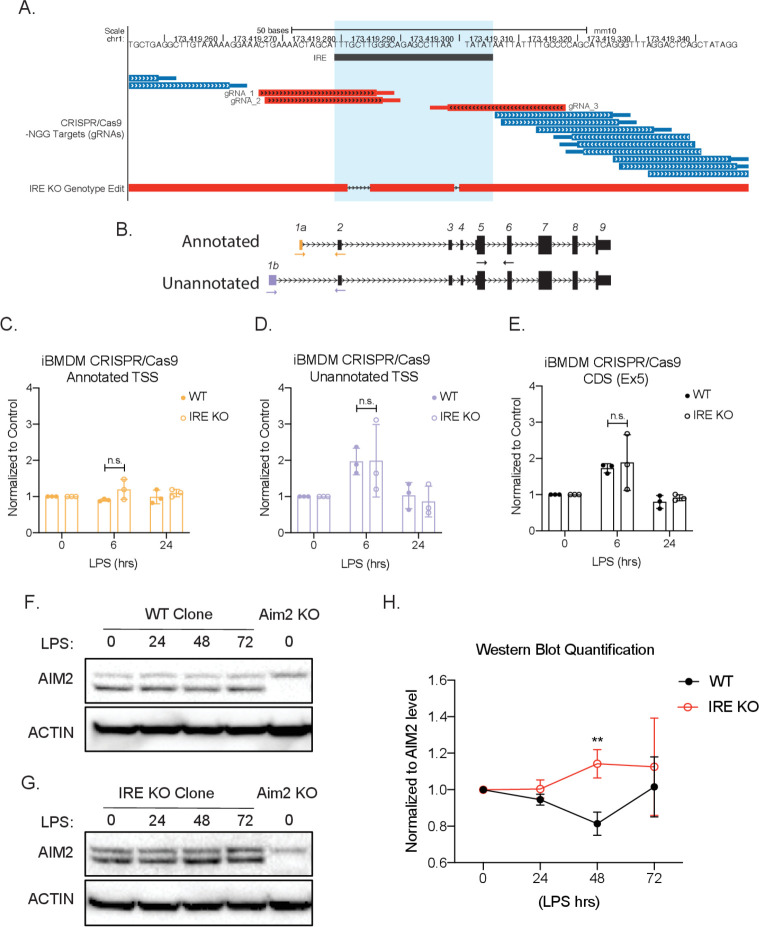
Determining the layers of gene regulation within the innate immune response is critical to our understanding of the cellular responses to infection and dysregulation in disease. We identified a conserved mechanism of gene regulation in human and mouse via changes in alternative first exon (AFE) usage following inflammation, resulting in changes to the isoforms produced. Of these AFE events, we identified 95 unannotated transcription start sites in mice using a de novo transcriptome generated by long-read native RNA-sequencing, one of which is in the cytosolic receptor for dsDNA and known inflammatory inducible gene, Aim2. We show that this unannotated AFE isoform of Aim2 is the predominant isoform expressed during inflammation and contains an iron-responsive element in its 5'UTR enabling mRNA translation to be regulated by iron levels. This work highlights the importance of examining alternative isoform changes and translational regulation in the innate immune response and uncovers novel regulatory mechanisms of Aim2.
Keywords: Macrophages; genetics; genomics; human; immunology; inflammation; mouse; splicing.
© 2021, Robinson et al.
Conflict of interest statement
ER, PJ, SC, MC, VS, RS, BS, RA, MJ, SC, SC No competing interests declared, MA holds options in Oxford Nanopore Technologies (ONT), is a paid consultant to ONT, received reimbursement for travel, accommodation and conference fees to speak at events organized by ONT,received research funding from ONT and is an inventor on 11 UC patents licensed to ONT (6,267,872, 6,465,193, 6,746,594, 6,936,433, 7,060,50, 8,500,982, 8,679,747, 9,481,908, 9,797,013, 10,059,988, and 10,081,835), AB received reimbursement for travel, accommodation and conference fees to speak at events organized by Oxford Nanopore Technologies (ONT)
Figures
Similar articles
-
Global Transcriptomics Uncovers Distinct Contributions From Splicing Regulatory Proteins to the Macrophage Innate Immune Response.Front Immunol. 2021 Jul 9;12:656885. doi: 10.3389/fimmu.2021.656885. eCollection 2021. Front Immunol. 2021. PMID: 34305890 Free PMC article.
-
Bcrp1 transcription in mouse testis is controlled by a promoter upstream of a novel first exon (E1U) regulated by steroidogenic factor-1.Biochim Biophys Acta. 2013 Dec;1829(12):1288-99. doi: 10.1016/j.bbagrm.2013.10.008. Epub 2013 Nov 2. Biochim Biophys Acta. 2013. PMID: 24189494 Free PMC article.
-
Functional characterization of two novel 5' untranslated exons reveals a complex regulation of NOD2 protein expression.BMC Genomics. 2007 Dec 20;8:472. doi: 10.1186/1471-2164-8-472. BMC Genomics. 2007. PMID: 18096043 Free PMC article.
-
Tissue specific glucocorticoid receptor expression, a role for alternative first exon usage?Biochem Pharmacol. 2006 Nov 30;72(11):1529-37. doi: 10.1016/j.bcp.2006.07.005. Epub 2006 Aug 22. Biochem Pharmacol. 2006. PMID: 16930562 Review.
-
Epigenetic Mechanisms Governing Innate Inflammatory Responses.J Interferon Cytokine Res. 2016 Jul;36(7):454-61. doi: 10.1089/jir.2016.0003. J Interferon Cytokine Res. 2016. PMID: 27379867 Free PMC article. Review.
Cited by
-
Widespread occurrence of hybrid internal-terminal exons in human transcriptomes.Sci Adv. 2022 Jan 21;8(3):eabk1752. doi: 10.1126/sciadv.abk1752. Epub 2022 Jan 19. Sci Adv. 2022. PMID: 35044812 Free PMC article.
-
Taking AIM at Influenza: The Role of the AIM2 Inflammasome.Viruses. 2024 Sep 27;16(10):1535. doi: 10.3390/v16101535. Viruses. 2024. PMID: 39459869 Free PMC article. Review.
-
CRISPRi screen uncovers lncRNA regulators of human monocyte growth.J Biol Chem. 2025 Jun;301(6):110204. doi: 10.1016/j.jbc.2025.110204. Epub 2025 May 7. J Biol Chem. 2025. PMID: 40345583 Free PMC article.
-
Generation of an isoform-level transcriptome atlas of macrophage activation.J Biol Chem. 2021 Jan-Jun;296:100784. doi: 10.1016/j.jbc.2021.100784. Epub 2021 May 14. J Biol Chem. 2021. PMID: 34000296 Free PMC article.
-
MAEL gene contributes to bovine testicular development through the m5C-mediated splicing.iScience. 2023 Jan 6;26(2):105941. doi: 10.1016/j.isci.2023.105941. eCollection 2023 Feb 17. iScience. 2023. PMID: 36711243 Free PMC article.
References
-
- Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, Bhatta A, Schattgen SA, McGowan JD, Blin J, Braun JE, Gandhi P, Moore MJ, Chang HY, Lodish HF, Caffrey DR, Fitzgerald KA. A long noncoding RNA Lincrna-eps acts as a transcriptional brake to restrain inflammation. Cell. 2016;165:1672–1685. doi: 10.1016/j.cell.2016.05.075. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
