

A phylogenetic approach for weighting genetic sequences
- PMID: 34049487
- PMCID: PMC8164272
- DOI: 10.1186/s12859-021-04183-8
A phylogenetic approach for weighting genetic sequences
Abstract
Background: Many important applications in bioinformatics, including sequence alignment and protein family profiling, employ sequence weighting schemes to mitigate the effects of non-independence of homologous sequences and under- or over-representation of certain taxa in a dataset. These schemes aim to assign high weights to sequences that are 'novel' compared to the others in the same dataset, and low weights to sequences that are over-represented.
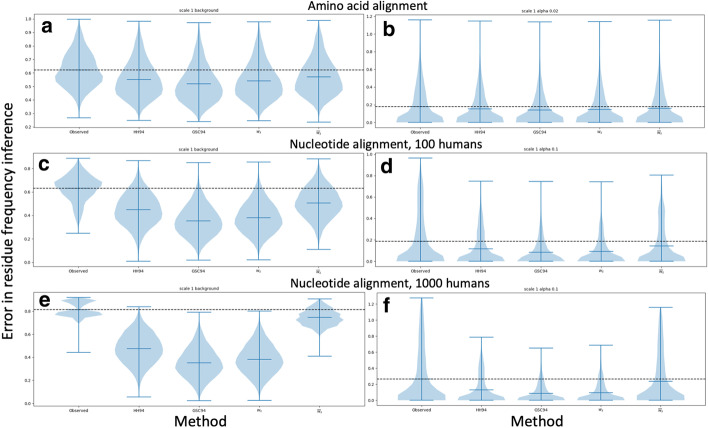
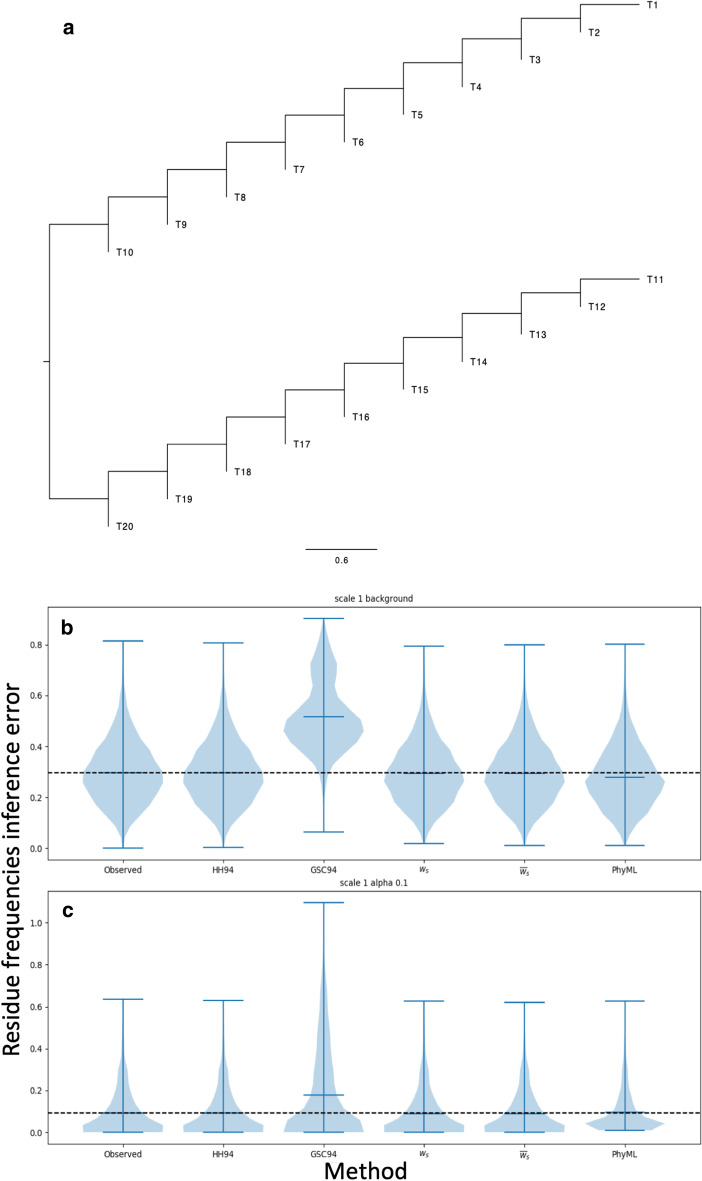
Results: We formalise this principle by rigorously defining the evolutionary 'novelty' of a sequence within an alignment. This results in new sequence weights that we call 'phylogenetic novelty scores'. These scores have various desirable properties, and we showcase their use by considering, as an example application, the inference of character frequencies at an alignment column-important, for example, in protein family profiling. We give computationally efficient algorithms for calculating our scores and, using simulations, show that they are versatile and can improve the accuracy of character frequency estimation compared to existing sequence weighting schemes.
Conclusions: Our phylogenetic novelty scores can be useful when an evolutionarily meaningful system for adjusting for uneven taxon sampling is desired. They have numerous possible applications, including estimation of evolutionary conservation scores and sequence logos, identification of targets in conservation biology, and improving and measuring sequence alignment accuracy.
Keywords: Alignment; Conservation scores; Phylogenetics; Protein profile; Sequence weights.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
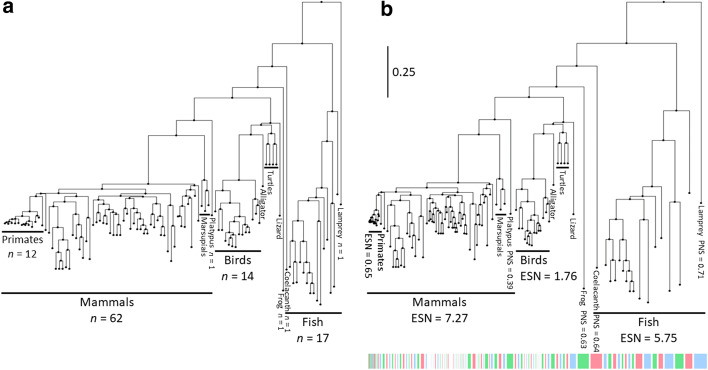
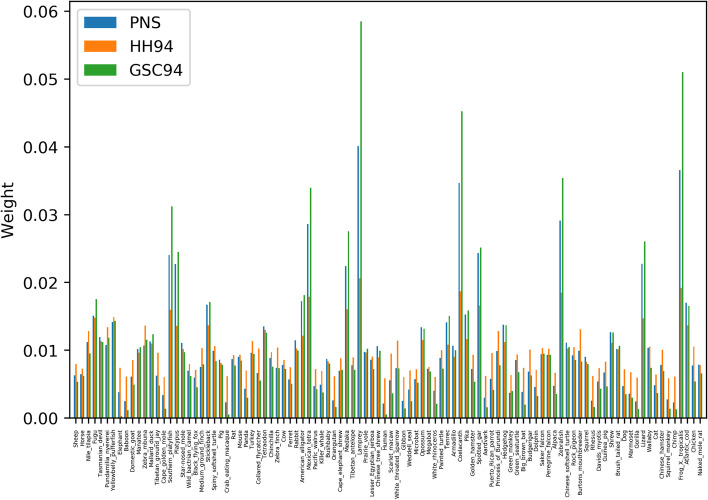
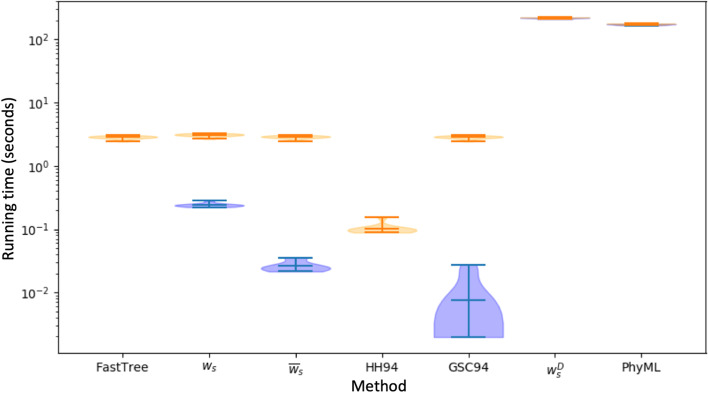
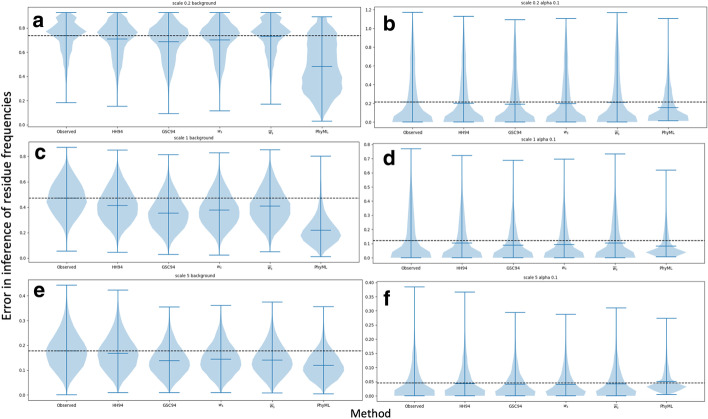
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
