

R-Loop-Mediated ssDNA Breaks Accumulate Following Short-Term Exposure to the HDAC Inhibitor Romidepsin
- PMID: 34050002
- PMCID: PMC8974437
- DOI: 10.1158/1541-7786.MCR-20-0833
R-Loop-Mediated ssDNA Breaks Accumulate Following Short-Term Exposure to the HDAC Inhibitor Romidepsin
Abstract
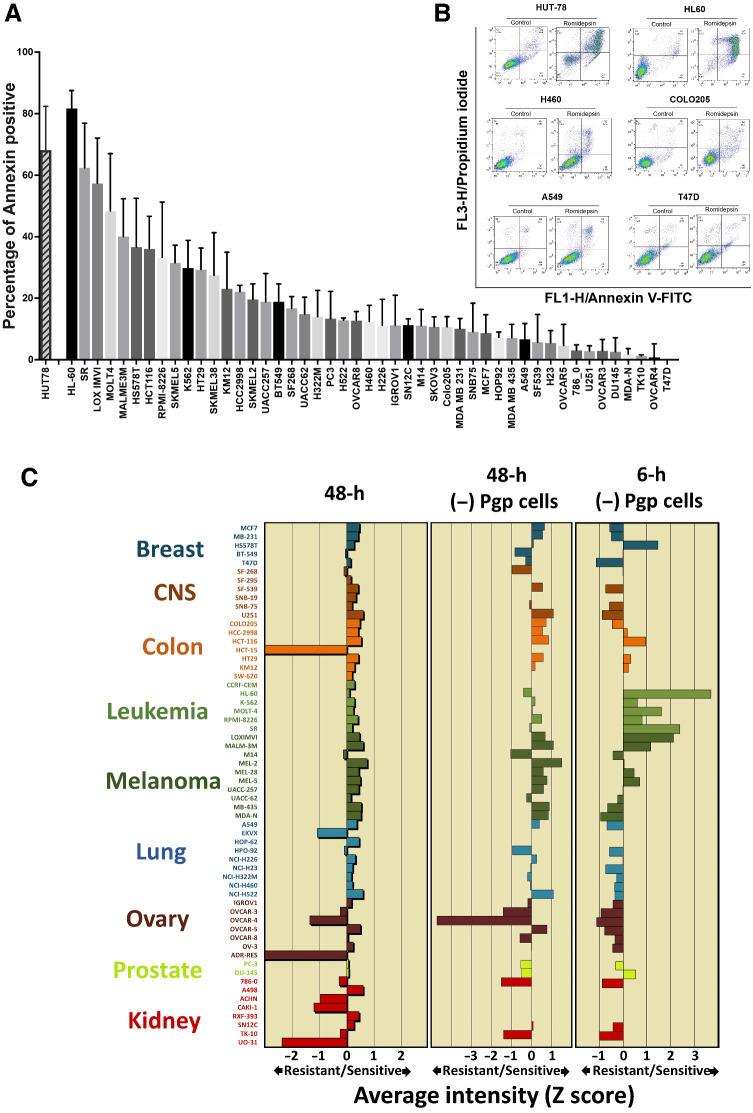
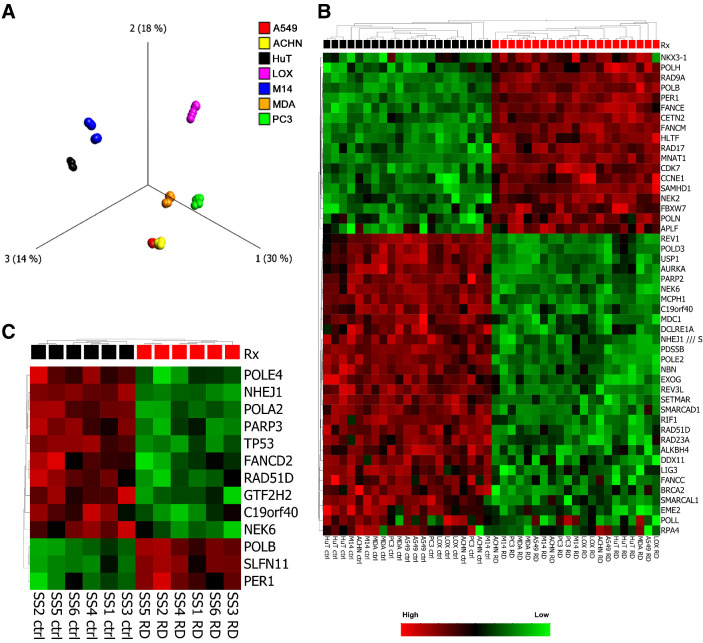
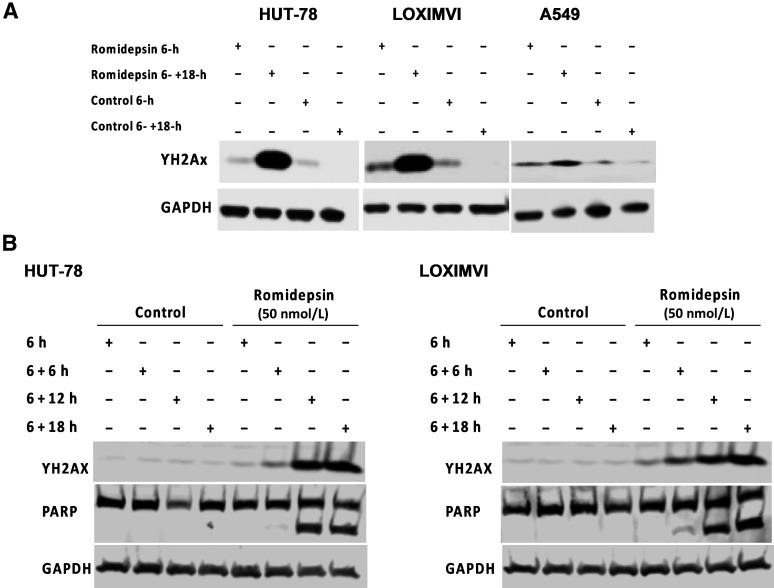
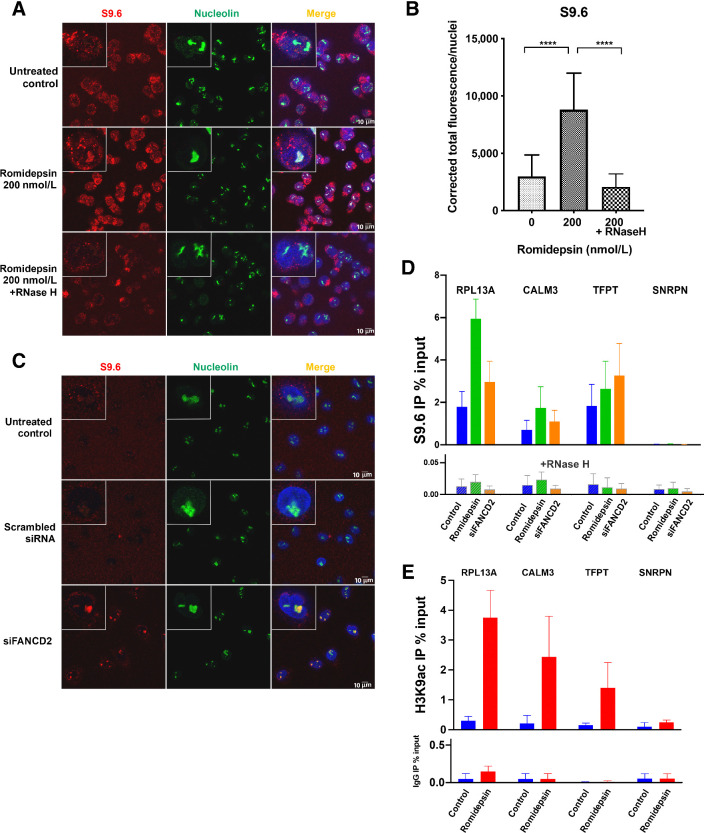
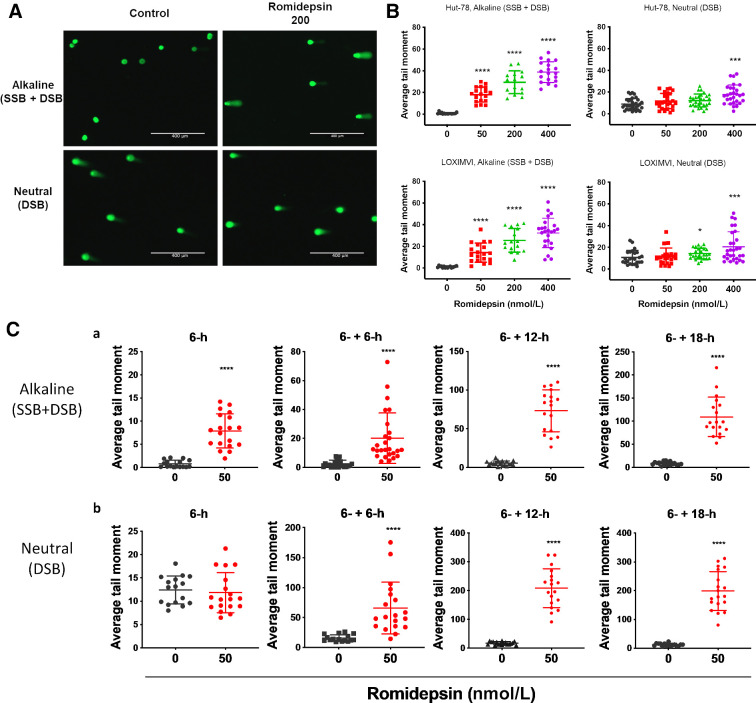
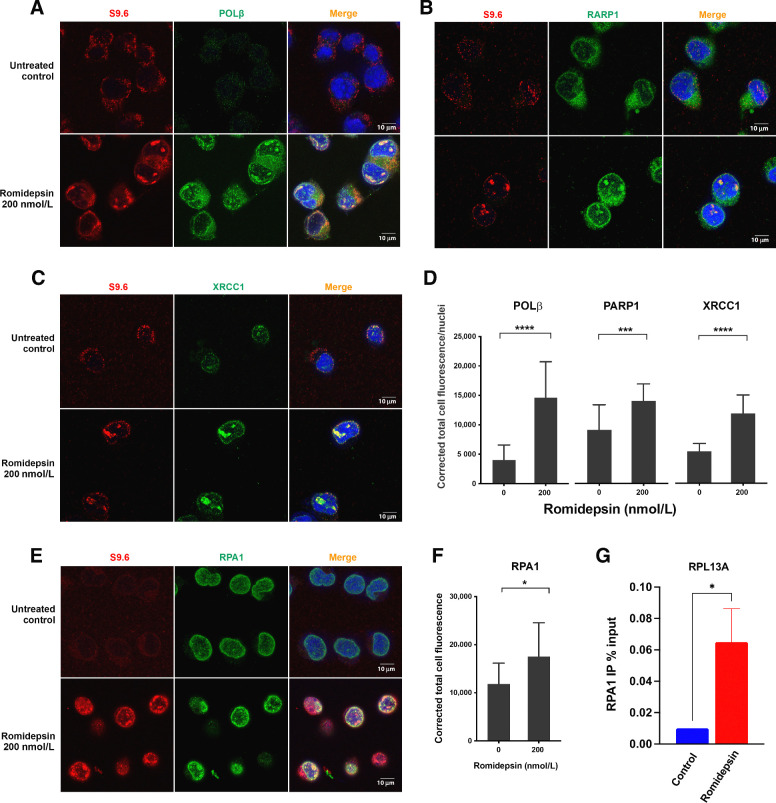
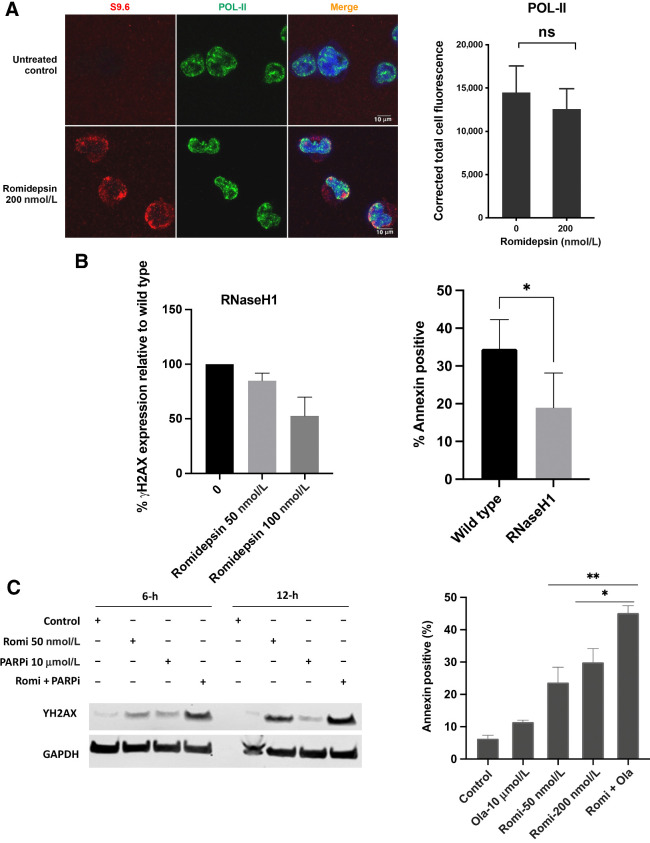
Histone deacetylase inhibitors (HDACi) induce hyperacetylation of histones by blocking HDAC catalytic sites. Despite regulatory approvals in hematological malignancies, limited solid tumor clinical activity has constrained their potential, arguing for better understanding of mechanisms of action (MOA). Multiple activities of HDACis have been demonstrated, dependent on cell context, beyond the canonical induction of gene expression. Here, using a clinically relevant exposure duration, we established DNA damage as the dominant signature using the NCI-60 cell line database and then focused on the mechanism by which hyperacetylation induces DNA damage. We identified accumulation of DNA-RNA hybrids (R-loops) following romidepsin-induced histone hyperacetylation, with single-stranded DNA (ssDNA) breaks detected by single-cell electrophoresis. Our data suggest that transcription-coupled base excision repair (BER) is involved in resolving ssDNA breaks that, when overwhelmed, evolve to lethal dsDNA breaks. We show that inhibition of BER proteins such as PARP will increase dsDNA breaks in this context. These studies establish accumulation of R-loops as a consequence of romidepsin-mediated histone hyperacetylation. We believe that the insights provided will inform design of more effective combination therapy with HDACis for treatment of solid tumors. IMPLICATIONS: Key HDAC inhibitor mechanisms of action remain unknown; we identify accumulation of DNA-RNA hybrids (R-loops) due to chromatin hyperacetylation that provokes single-stranded DNA damage as a first step toward cell death.
©2021 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
T. Litman reports personal fees from LEO Pharma A/S outside the submitted work. A. Basseville reports grants from the European Commission during the conduct of the study. O.A. O'Connor reports grants from Celgene outside the submitted work. S.E. Bates reports grants from Celgene during the conduct of the study; as well as a patent for Depsipeptide for therapy of kidney cancer issued and Deacetylase inhibitor therapy issued. No disclosures were reported by the other authors.
Figures
Similar articles
-
Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir.Int J Cancer. 2016 Jan 1;138(1):125-36. doi: 10.1002/ijc.29698. Epub 2015 Aug 11. Int J Cancer. 2016. PMID: 26205347
-
The Histone Deacetylase Inhibitor Romidepsin Spares Normal Tissues While Acting as an Effective Radiosensitizer in Bladder Tumors in Vivo.Int J Radiat Oncol Biol Phys. 2020 May 1;107(1):212-221. doi: 10.1016/j.ijrobp.2020.01.015. Epub 2020 Jan 25. Int J Radiat Oncol Biol Phys. 2020. PMID: 31987970 Free PMC article.
-
Romidepsin induces cell cycle arrest, apoptosis, histone hyperacetylation and reduces matrix metalloproteinases 2 and 9 expression in bortezomib sensitized non-small cell lung cancer cells.Biomed Pharmacother. 2014 Apr;68(3):327-34. doi: 10.1016/j.biopha.2014.01.002. Epub 2014 Jan 15. Biomed Pharmacother. 2014. PMID: 24485799
-
Molecular mechanistic pathways underlying the anticancer therapeutic efficiency of romidepsin.Biomed Pharmacother. 2023 Aug;164:114774. doi: 10.1016/j.biopha.2023.114774. Epub 2023 May 22. Biomed Pharmacother. 2023. PMID: 37224749 Review.
-
Interstrand Crosslink Repair as a Target for HDAC Inhibition.Trends Pharmacol Sci. 2017 Sep;38(9):822-836. doi: 10.1016/j.tips.2017.05.009. Epub 2017 Jul 4. Trends Pharmacol Sci. 2017. PMID: 28687272 Review.
Cited by
-
NOXA Accentuates Apoptosis Induction by a Novel Histone Deacetylase Inhibitor.Cancers (Basel). 2023 Jul 17;15(14):3650. doi: 10.3390/cancers15143650. Cancers (Basel). 2023. PMID: 37509312 Free PMC article.
-
Mechanisms underlining R-loop biology and implications for human disease.Front Cell Dev Biol. 2025 Feb 21;13:1537731. doi: 10.3389/fcell.2025.1537731. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40061014 Free PMC article. Review.
-
Looping forward: exploring R-loop processing and therapeutic potential.FEBS Lett. 2025 Jan;599(2):244-266. doi: 10.1002/1873-3468.14947. Epub 2024 Jun 6. FEBS Lett. 2025. PMID: 38844597 Free PMC article. Review.
-
R-Loops and R-Loop-Binding Proteins in Cancer Progression and Drug Resistance.Int J Mol Sci. 2023 Apr 11;24(8):7064. doi: 10.3390/ijms24087064. Int J Mol Sci. 2023. PMID: 37108225 Free PMC article. Review.
-
M6A-methylated circPOLR2B forms an R-loop and regulates the biological behavior of glioma stem cells through positive feedback loops.Cell Death Dis. 2024 Aug 1;15(8):554. doi: 10.1038/s41419-024-06946-6. Cell Death Dis. 2024. PMID: 39090090 Free PMC article.
References
-
- Leder A, Leder P. Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell 1975;5:319–22. - PubMed
-
- Mickley LA, Bates SE, Richert ND, Currier S, Tanaka S, Foss F, et al. Modulation of the expression of a multidrug resistance gene (mdr-1/P-glycoprotein) by differentiating agents. J Biol Chem 1989;264:18031–40. - PubMed
-
- Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na+/I− symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metabol 2001;86:3430–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
